شیمی - بازرسی فنی
وبلاگ هر هفته یک حدیث به آدرس www.hadis89.blogsky.com منتظر مشتاقان احادیث پیامبر و ائمه معصومین (ع) می باشد.شیمی - بازرسی فنی
وبلاگ هر هفته یک حدیث به آدرس www.hadis89.blogsky.com منتظر مشتاقان احادیث پیامبر و ائمه معصومین (ع) می باشد.تفلون چیست؟
تفلون نام تجاری "پلی تترافلوئورواتیلن"، همان محصول چند میلیارد دلاری شرکت دوپون است که در موارد گوناگونی، از ماهیتابه های نچسب گرفته تا لباسهای فضایی و دریچه های مصنوعی قلب، استفاده شده است. کشف آن ناشی از تصادفی بود که "روی ج. پلانکت" ( Roy.J. Plunkett) ، شیمیدان جوانی در شرکت دوپون که تنها دو سال قبل از روز سرنوشت ساز 6 آوریل سال 1938 ، دکترای خود را از دانشگاه ایالتی اوهایو گرفته بود، مشاهده کرد. در این روز دکتر پلانکت مخزنی از "تترافلوئورَ واتیلن گازی" ، باز کرد به این امید که سرد کننده ای غیر سمّی از آن تهیه کند. اما پلانکت و دستیارش جک ریبوک با شگفتی دیدند که گازی خارج نشد. پلانکت نمی توانست این پدیده را توجیه کند، چون وزن مخزن نشان می داد که باید پر از "فلوئوروکربن گازی" باشد.
پلانکت تصمیم گرفت به جای آنکه برای ادامه پژوهش در زمینه مواد سرد کننده ، مخزن را دور بیندازد و مخزن جدیدی بگیرد، کنجکاوی اش را در باره آن مخزن " خالی" ارضاء کند. وقتی سیمی به دریچه مخزن وارد کرد و مطمئن شد که اشکالی ندارد، مخزن را اره کرد و به درونش نگاهی انداخت. در آنجا گرد سفید مومی شکلی پیدا کرد و چون شیمیدان بود، فهمید که ممکن است این مشاهده چه معنایی داشته باشد.
مولکولهای تترافلوئورواتیلن گازی به حدی با یکدیگر ترکیب ( پلیمریزه) شده بودند که ماده جامدی تشکیل دادند. هیچ کس تا آن هنگام پلیمریزاسیون این ترکیب بخصوص را مشاهده نکرده بود، اما با این حال واکنش به نحوی در مخزن "خالی" مرموز صورت گرفته بود. چندی نگذشت که این کشف تصادفی و ویژگی های عجیب پلیمر به دست آمده، پلانکت و دیگر شیمیدانان شرکت دوپون را واداشت تا راههایی پیدا کنند که "پلی تترافلوئور واتیلن" را برحسب نیاز تولید کنند.
واقعاً هم که این گرد سفید مومی شکل ویژگیهای عجیبی داشت: از شن هم خنثی تر بود. نه تحت تأثیر اسیدها و بازی های قوی قرار می گرفت، نه حرارت. هیچ حلّالی هم آن را حل نمی کرد اما بر خلاف شن بسیار" لیز" بود. با وجود این ویژگی های جالب وغیرعادی، اگر جنگ جهانی دوم در نگرفته بود، چه بسا به دلیل گرانی این پلیمر جدید، تا مدتها بعد کار دیگری در زمینه آن صورت نمی گرفت. اما چند ماهی نگذشته بود که دانشمندانی که مشغول ساختن نخستین بمب اتمی بودند ، احیتاج به ماده ای پیدا کردند تا بتوانند از آن واشرهایی بسازند که در برابر گازِ بسیار خورنده هگزافلوئورید اورانیم، که برای تولید اورانیم 235 بمب مصرف می شد، مقاوم باشد.
از قضا سرهنگ لزلی ر. گرووز، مسئول بخش طرح بمب اتمی در ارتش ایالات متحده، از طریق آشنایانی که در شرکت دوپون داشت از پلاستیک جدیدشان که فوق العاده خنثی بود، خبردار شد. وقتی به گرووز گفته شد ممکن است این پلاستیک جدید گران تمام شود، پاسخ داد که در این طرح ، قیمت به هیچ وجه مطرح نیست. بدین ترتیب این پلیمر لغزنده در واشرها و دریچه ها به کار رفت، و واقعاً هم نسبت به ترکیب خورنده اورانیم مقاوم بود. شرکت دوپون در طی جنگ، تفلون را برای این کاربرد تولید کرد و عموم مردم تا بعد از جنگ هم چیزی درباره این پلیمر جدید نمی دانستند.
در واقع در سال 1960 بود که نخستین ماهیتابه ها و ظروف شیرینی پزی پوشیده از تفلون به بازار آمدند. این فرآورده های تفلونی مانند بسیاری از محصولات پلیمری جدید موقعی که نخستین بار به مردم معرفی شدند، چندان نتایج امیدوار کننده ای نداشتند. گرچه این پلاستیک به عنوان یک سطح خوراکپزی نچسب بسیار مناسب بود، اما به سختی به ظروف فلزی پیوند می شد، بنابراین در برابر شست و شوی زنان خانه داری که عادت داشتند دیگ و ماهیتابه هایشان را محکم بسابند، مقاوم نبود. پس از آنکه روشهای گوناگونی امتحان شدند و چهار نسل پوشش تفلونی به تولید رسیدند، دوپون در سال 1986 اعلام کرد سیلورستون سوپرای آنان دو برابر مقاومتر از نسل سوم سیلورستون است. در همین ضمن کاربردهای متعدد دیگری کشف شده بودند که دیگر پوشاندن ظروف خوراکپزی را نسبتاً بی اهمیت جلوه می دادند.
تفلون به طرق گوناگون بر زندگی میلیونها نفر در سراسر جهان داشته است.پلانکت می گوید آن قدر کسانی که ضربانساز یا سرخرگ آئورت تفلونی در بدنشان تعبیه شده و امروز جانشان نجات یافته است برایش نامه می فرستند و تلفن می کنند که به قول خودش نمی تواند از پس آنها برآید. چون تفلون از معدود موادی است که بدن ، آن را در هنگام پیوند رد نمی کند. از آن می توان در ساخت قرنیه های مصنوعی، استخوانهای جایگزین برای چانه، بینی، جمجمه، مفاصل ران و زانو، قطعات گوش، نای مصنوعی، دریچه های قلب، زرد پی ها، بخیه ها، مجاری صفراوی و دندانهای مصنوعی، استفاده کرد.
از تفلون در پوشش بیرونی لباسهای فضانوردان استفاده شده است. تفلون ماده عایق کننده سیمها و کابلهای برقی است که در برابر تابش شدید خورشید بر سطح ماه مقاومت کرده اند. مخروطه دماغه و دیگر سپرهای گرمایی سفینه های فضایی و نیز مخازن سوخت آنها از تفلون ساخته شده اند.
همه این کاربردهای مهم و ارزشمند، ثمره کشف بخت یارانه روی پلانکت بوده اند. آری، تصادفی بیش نبود، اما فقط به سبب کنجکاوی و ذکاوت مردی که این تصادف برایش اتفاق افتاد، به اکتشافی تبدیل شد.
اشاره
آنچه باعث شد اصلاً وظیفه ترکیب سرد کننده ای از فلوئور به روی پلانکت داده شود، خود اتفاق بخت یارانه دیگری بود. در سال 1928، "چارلزف. کترینگ" ( ملقب به "رئیس") از بخش مواد سرد کننده جنرال موتورز، جست وجو به دنبال سرد کننده بی خطری را آغاز کرد- ترکیبی که بی رنگ، بی بو، بی طعم، غیرسمّی و غیر آتشگیر باشد، تا جایگزین مواد سمّی و زیانمندی نظیر آمونیاک و دی اکسید گوگرد که در آن هنگام در یخچالها استفاده می شدند بشود. "توماس میجلی" و "آلبرت هن" پس از بررسی دقیق منابع و مراجع شیمی نتیجه گرفتند که گرچه گاه گزارش شده بود ترکیبات فلوئور سمّی اند، اما شاید برخی از ترکیبات فلوئوروکلردارکربن مناسب باشند.
برای آنکه میجلی و هن این گزارشها را تایید کنند، لازم بود نمونه هایی از کلروفلوئوروکربن های ساده تهیه و آنها را در آزمایش های جانوری امتحان کنند. آنان از یکی از انبارهای مواد شیمیایی تقاضای پنج بطری 30 گرمی تری فلوئورید آنتیموان کردند ( یعنی تمام ذخیره ای که از این ماده شیمیایی در ایالات متحده وجود داشت!). یکی از این پنج بطری را به طور اتفاقی برگزیدند و از آن برای تهیه کلروفلوئوروکربن استفاده کردند. یک خوکچه هندی را زیر ظرفی شیشه ای که ترکیب کلروفلوئوروکربن گازی در آن بود گذاشتند و دیدند که جانور به هیچ وجه تحت تأثیر گاز قرار نگرفت. این مشاهده گمان آنان را مبنی بر سمّی نبودن ترکیبات آلی فلوئوردار اثبات کرد.
شیمیدانان برای تأیید این آزمایش نمونه های دیگری از گاز کلروفلوئوروکربن را با استفاده از بقیه بطری های تری فلوئورید آنتیموان تهیه و همین آزمایش را روی خوکچه های هندی تکرار کردند. در همه این آزمایشها خوکچه ها مردند! بررسی دقیق تر نشان داد که در تمام بطری های تری فلوئورید آنتیموان جز یکی آب وجود داشت. آبی که در چهار پنجم نمونه ها بود منجر به تولید گاز مرگبار فسژن شد (کلرفسژن از کلرید آلی استفاده شده به همراه تری فلوئورید آنتیموان که ماده اولیه تهیه کلروفلوئوروکربن بود تأمین شد). بنابراین علت مرگ جانوران وجود فسژن بود.
اگر میجلی و هن تصادفاً در نخستین آزمایش جانوریشان شیشه ی تری فلوئورید آنتیموان خشک را انتخاب نمی کردند، چه بسا خیال استفاده از کلروفلوئوروکربن ها را به عنوان مواد سرد کننده ای از سر به در می کردند و گزارش های ( نادرست) قبلی را مبنی بر سمّی بودن این ترکیبات می پذیرفتند. اما سرمایه گذاری مشترک شرکت های جنرال موتورز و دوپون منجر به تأسیس بخش فرئون دردوپون برای تحقیق و توسعه شیمی کلروفلوروکربنها شد، و در آنجا بود که روی پلانکت تفلون را کشف کرد.
واژگان شیمی تجزیه
| معادل فارسی | تعریف | واژه لاتین |
| اسید سیستم حلالی | ماده ای که کاتیون حلال را میدهد. | salvent-system acid |
| باز سیستم حلالی | ماده ای که آنیون حلال را میدهد | solvent-system base |
| ماده آمفی پروتیک | ماده ای که میتواند به صورت یک اسید برونشتد عمل کند. | amphiprotic substance |
| خنثی کردن (شدن) | واکنش بین یک اسید و یا باز | neutralization |
| آبکافت | واکنش یک کاتیون یا یک آنیون با آب که PH را تحت تاثیر قرار میدهد. | hydrolysis |
| اثر یون مشترک | اثر ناشی از افزایش یک ترکیب روی یک سیستم در حال تعادل به طوری که ترکیب با ترکیب حاضر در سیستم ، در یک یون ، مشترک باشند. | common ion effect |
| ثابت تفکیکی آب | حاصلضرب غلظت +H و غلظت -OH در هر سیستم آبی در ۲۵ درجه سانتیگراد | dissociation constant of water |
| ثابت تفکیک اسید | ثابت تعادلی که به تعادل شامل یک اسید ضعیف و یونهای مشتق از آن در محلول آبی مربوط میشود. | dissociation constant of acid |
| ثابت تفکیک باز | ثابت تعادلی که به تعادل شامل یک باز ضعیف و یونهای مشتق از آن در محلول آبی مربوط میشود. | dissociation constant of base |
| درجه تفکیک | کسری از غلظت کل الکترولیت ضعیفی که در محلول آبی و در حال تعادل به شکل یونی باشد. | degree of dissociation |
| شناساگر اسید- باز | ترکیبی که با تغییر PH محلول ، رنگ آن تغییر میکند. | acid –base indicator |
| معادله هندرسون – هاسل باخ | معادله ای که محاسبه PH را میسر میسازد. {PH=PKa+Cog-{A-}/{HA که در آن ، PKa لگارییم منفی ثابت تفکیک اسید ضعیفی است که برای تهیه بافر مورد استفاده قرار گرفته است. {-A} غلظت یونها و {HA} غلظت مولکولهای اسید ضعیف است. | Hendrerson Hasselbalch equation |
| منحنی تیتراسیون | نموداری که نشان میدهد چگونه PH یک محلول در حین عمل تیتراسیون تغییر مییابد. PH در برابر حجم باز یا اسید اضافه شده رسم میگردد. | Titration Curve |
| نقطه پایانی | نقطه ای در تیتراسیون که شناساگر ، تغییر رنگ مییابد. | end point |
| نقطه همارزی | نقطه ای در تیتراسیون که مقدار همارزی از باز یا اسید به نمونه ای اسید یا بازی که تیتر میشود، اضافه میگردد. | equivalence point |
| PH | لگاریتم منفی (بر مبنای ۱۰) غلظت یونهای +H در محلول آبی | PH |
| PK | لگاریتم منفی (بر مبنای ۱۰) ثابت تعادل | PK |
| POH | لگاریتم منفی (بر مبنای ۱۰) غلظت یونهای -OH در محلول آبی | POH |
| آمفوتریسم | خاصیت هیدروکسید بعضی از فلزات که میتوانند هم به عنوان اسید عمل کنند و هم به عنوان باز. مواد آمفوتر در آب نامحلولند. | amphoterism |
| اثر نمک | افزایش انحلال پذیری موادیکه کم محلولند. این افزایش به دنبال افزایش الکترولیت دیگر به محلول مشاهده میشود. | salt effect |
| ثابت ناپایداری | ثابت تعادل برای تفکیکی کامل یون کمپلکس به کاتیون فلزی و لیگاند. عکس این ثابت ، ثابت تشکیل نامیده میشود. | instability constant |
| حاصلضرب انحلال پذیری | ثابت تعادل برای سیستمی را گویند که شامل ماده ای کممحلول و در حال تعادل با محلول سیر شده نمکهایش است. این مقدار ثابت برابر حاصلضرب غلظت یونها است، بهطوری که غلظتها به توان عددی که همان ضرایب معادله شیمیایی موازنه شده است، رسیده باشند. | solubility product |
| حاصلضرب یونی | مقداری است که از جایگزینی غلظتهای پیشنهاد شده در عبارتی مشابه عبارت حاصلضرب انحلال پذیری بدست میآید. با مطابقت دادن غلظت یونها با مقادیر پیشنهادی ، مقدار حاصلضرب یونی با مقدار ksp مقایسه میشود تا معلوم شود رسوب تشکیل میگردد یا نه. اگر حاصلضرب یونی از ksp باشد، رسوب تشکیل خواهد شد. | Ionic product |
| مول | مقدار ماده خالصی که عده واحدهای مستقل اصلی آن ، دقیقا برابر عده اتمهای موجود در ۱۲g کربن ۱۲ است. مجموعه ای که شامل عدد آووگادرو واحد مستقل باشد. | mole |
| حلال | جزیی از یک محلول که بیشترین مقدار موجود در محلول را دارد یا حالت فیزیکی محلول را مشخص میکند. | solvent |
| غلظت | مقدار ماده حل شده در مقدار معینی از محلول یا حلال | Concetration |
| ماده حل شده | ماده حل شده در حلال جزیی از یک محلول یا حلال. | solved substance |
| محصول | ماده ای که در یک واکنش شیمایی تولید میشود. | Product |
| معادله شیمیایی | نمایش یک واکنش شیمیایی با نمادها و فرمولهای عناصر و مواد مرکبی که در آن واکنش دخالت دارند. | Chemical equation |
| مولاریته | عده مولهای ماده حل شده در یک لیتر محلول | molarity |
| واکنش دهنده | ماده ای که در واکنش شیمیایی مصرف میشود. | reactant |
| تیتراسیون | فرایندی که در آن ، یک محلول استاندارد با محلولی با غلظت نامعلوم واکنش داده میشود تا غلظت محلول مجهول تعیین شود. | titration |
| رسوب کردن | تشکیل یک ماده نامحلول (موسوم به رسوب) در مخلوط یک واکنش به حالت محلول | precipitate |
| محلول استاندارد | مخلولی که غلظت ماده حل شده در آن ، دقیقا معین است. | Standard solution |
| نرمالیته | عده اکی والانهای جسم حل شده در یک لیتر محلول | normality |
| نمک | ترکیبی که از واکنش یک اسید و یک باز بدست میآید. این ترکیب شامل یک کاتیون از باز و یک آنیون از اسید است. | salt |
| نمک اسیدی | نمکی که از خنثی شدن ناکامل یک اسید چند پروتونی بدست میآید. آنیونهای این نوع نمکها ، یک یا چند هیدروژن یونش پذیر اسید اولیه را نگه داشتهاند. | acidic salt |
| نمک خنثی | نمک حاصل از خنثی شدن کامل یک اسید چند پروتونی که در آن ، تمام پروتونهای اسید خنثی شده است. | neutral salt |
| وزن همارز | مقداری معین از هر ماده که بر اساس واکنش مورد بررسی ، به گونه ای محاسبه میشود که یک همارز از یک واکنش دهنده دقیقا با یک همارز از واکنش دهنده دیگر واکنش دهد. | equivalent weight |
| یون هیدرونیوم | یونی که از یک پروتون و یک مولکول آب تشکیل میشود و دارای فرمول +H۳O است. | Hydronium ion |
| بهره نظری | بیشترین مقدار محصولی که میتوان از یک معادله شیمیایی بدست آورد و محاسبه آن با استفاده از نظریه استوکیومتری بر مبنای معادله شیمیایی واکنش باشد. | Theoretical yield |
| بهره درصدی | بهره واقعی تقسیم بر بهره نظری ضرب در ۱۰۰ | percent yield |
| بهره واقعی | مقدار محصول بدست آمده از یک واکنش شیمیایی | actual yield |
اسید فسفریک
فسفریک اسید از جمله پرمصرف ترین مواد شیمیایی در صنعت است. به عنوان ماده افزودنی در نوشابه های گازدار کاربرد دارد و در تولید کودهای شیمیایی، پاک کننده های صابونی و غیر صابونی،تصفیه آب،خوراک دام و دارو سازی، مکملهای غذای دام و طیور(دی و منو کلسیم فسفات) ، مواد فسفاته شوینده ها ،تصفیه پسابها ، تولید کودهای فسفاته(مهمترین)، ضد حریق کردن برخی سطوح و عوامل بازدارنده اشتعال، ونیز جهت تمیز کردن و جرم گیری سطوح فلزی به کار می رود. فسفریک اسید خوراکی را از افزودن آب به P4O10 می سازند. اسید فسفریک اسید ضعیفی است و در شرایط عادی و مدت زمان کوتاه آنقدر نمیتواند خطرساز باشد .

کاربرد در صنایع غذایی
از اسید فسفریک در تولید غذاهای اسیدی و نوشابه های گازدار مانند انواع کولاها استفاده میشود. بکارگیری این ماده سبب دادن طعم تندی به غذا شده، و از آنجا که ماده شیمیایی با تولید انبوه است، با قیمتی ارزان و حجمی فراوان در دسترس میباشد. همانطور که ذکر شد، قیمت پایین و حجم زیاد تولید این ماده، آنرا در مقایسه با طعم دهنده های طبیعی نظیر زنجبیل برای دادن طعم تندی، یا اسید سیتریک که از لیمو (lemon) و عصاره لیموترش (lime) که برای دادن طعم ترشی بکار میرود، در رتبه بالاتری قرار داده است.
کاربرد در مواد پاک کننده
در تولید پاک کننده ها اسید فسفریک برای نرم کردن آب بکار می رود.آب نرم بدون یونهای کلسیم (II) و منیزیم (II) که آب سخت را تشکیل می دهند،اگر ازبین نروند تشکیل آب سخت را می دهند که این یونها با صابون تشکیل رسوبات غیر قابل حل می دهند که سبب لکه بروی لباس ها در هنگام شستشو می شوند.نمکهای فسفات از اسید فسفریک بطور وسیع در پاک کننده هابعنوان(builder) بکار می رود.بیشتر گستره ترکیبات فسفر درمخلوط پاک کنندهای جامد است که سدیم تری پلی فسفات یکی از آنهاست.Na5P3O10 ،بعنوان نرم کننده آب ،سدیم تری پلی فسفات با کلسیم (II) و منیزیم (II) یوند برقرار می کندو تشکیل اجزاء محلول را می دهد که کمپلکس یا کلیت است.این کمپلکس ها از واکنش کلسیم (II) و منیزیم (II) با صابون ممانعت بعمل می آورندتا رسوب ایجاد نشود.
کاربرد در زدودن زنگ آهن
از اسید فسفریک میتوان مستقیماً برای زدودن زنگ آهن (اکسید آهن III) از ابزارهای آهنی یا فولادی و تبدیل آهن به فسفاتهای محلول در آب استفاده نمود. پس از زدودن زنگ آهن فسفات آهن تولید شده تبدیل به ترکیب فسفات آهن سیاه شده که خود به عنوان عامل جلوگیری از خوردگی میتواند مورد استفاده قرار گیرد. اسید فسفریک به عنوان کاتالیت در صنایع پتروشیمی کاربرد دارد.
کاربرد در پزشکی
از اسید فسفریک در دندانپزشکی و اورتودنسی بهعنوان عامل قلم زنی (Etching) جهت تمیز کردن و زبر کردن سطح دندان خصوصاً در جاهایی که از اسباب و وسایل دندانپزشکی استفاده شده، بکار میرود. همچنین از اسید فسفریک بهعنوان کاتالیست در ساخت آسپیرین بخاطر داشتن یون هیدروژن فراوان و آلایندگی کمتر در مقایسه با اسید کلریدریک و سولفوریک استفاده میشود.
کاربرد در کشاورزی
بیشتر اسید فسفریک در تولید کود بکار می رود.فسفر یکی از عناصر ضروری برای رشد گیاهان محسوب می شود.فسفاتهای آلی ترکیباتی هستند که انرژی لازم برای بیشتر واکنشهایی که در سلولهای زنده اتفاق می افتند را مهیا می کنند.بنابراین خاکهای غنی با کودهای فسفاتی رشد گیاهان را بالا می برند. افزایش غلظت فسفات در سطح آبها همچنین رشد گیاهان آبزی را بالا می برد.اضافات کودهای شیمیایی می توانند باعث تحریک رشد گیاهان در آبهای دریاچه ها و آبهای جاری بشوند.آب فاضلاب ها که شامل فسفات است می تواند تاثیر یکسانی داشته باشد.
دریاچه ها که از مواد مغذی غنی هستند از افزایش سرعت Eutrophication(انباشتگی خوراک آبی) تلف می شوند. وقتی گیاهان آبزی با این شرایط مغزی بودن محیط رشد می کنند پس از مدتی خواهند مرد. حال این گیاهان مرده برای تجزیه اکسیژن حل شده در آب را مصرف می کنند. این مصرف سطح اکسیژن حل شده را کاهش می دهد تا نقطه ای که برای حمایت حیوانات آبزی کافی نیست. برای کاهش تهدید Eutrophication دریاچه بسیاری از مناطق دارند فسفات ها را از پاک کننده ها حذف می کنند. در بعضی از موارد فسفاتها جای خودشان را با کربناتها عوض می کنند. به عبارت دیگر پاک کنندهای جدید به گونه ای تهیه می شوند که با کلسیم (II) و منیزیم (II) یونهای سخت آب واکنش ندهند.
روشهای تولید اسید فسفریک در صنعت
روش تر(Wet process): در اثر اضافه کردن اسید سولفوریک روی فسفات کلسیم بدست می آید. طبق واکنش زیر:

در این روش کانیهای فسفات را با اسید سولفوریک ترکیب می کنند، علاوه بر تولید اسید فسفریک و برخی فسفاتها که ترکیبات اصلی هستند، سایر ترکیبات (CACO3CAF2)هم تولید میشوند، از طرف دیگر واکنشهایی که در آن با از بین رفتن اسید سولفوریک ، ترکیباتی تولید میشود که از نظر تجاری کم اهمیت هستند (واکنشهای پارازیتی)، یونهای مزاحمی تولید شده و باعث آلودگی مخلوط ها میشوند .
روش حرارتی(خشک)Thermal process:
این روش شامل احتراق فسفر و هیدراسیون P4O10 میباشد. مخلوط فسفر مایع و هوا به محفظه احتراق که شبیه برج است تزریق شده و با انجام واکنش اکسیداسیون فسفر، تولید میشود. جنس محفظه احتراق نوعی فولاد مخصوص است که با H3PO4 غیرفعال شده است. P4O10 به دست آمده را در برج بعدی هیدراته میکنند و بخارات باقیمانده P4O10 را ، واحد شستشو به اسید فسفریک رقیق تبدیل کرده و به عنوان افشان در پایین آوردن دمای برج اول استفاده میشود.
روش های تولید در ایران
در ایران از اثر اسید سولفوریک بر کانیهای آپاتیتی اسید فسفریک تولید می شود . از آنجا که فسفر وآرسنیک در یک گروه(گروه 5) جدول شیمیایی (تناوبی) قرار دارند و در طبیعت نیز به احتمال زیاد در کانیهای مختلف با هم وجود دارند و فقط در درصد خلوص انها اختلاف می باشد.
چنانکه میدانید آرسنیک از عناصر سمی و کشنده به شمار می آید و وجود آن در کانیهای آپاتیتی که اسید فسفریک از آنها تهیه میشود و غالبا در صنایع خوراک دام و بهداشتی مصرف میگردد خطری مهم محسوب میگردد. البته روشهای تولید اسید فسفریک مثل احتراق فسفر خالص این مشکل را حل نموده اما قیمت تمام شده بسیار بالاتر از بهره گیری از کانی های آپاتیتی است. فلوئور یکی دیگر ازموادی است که سمیت دارند و جزء لاینفک کانی های آپاتیتی است.
تعیین مقدار اکسیژن محلول
مقدمه:
تمام موجودات زنده برای انجام متابولیسم و تهیه انرژی جهت رشد و تولید مثل نیاز به اکسیژن به فرمهای مختلف دارند. واکنشهای هوازی دارای اهمیت خاصی بوده زیرا به اکسیژن آزاد نیاز دارند. تمام گازخای موجود در هوا به مقدارهای مختلف در آب محلولند. نیتروژن و اکسیژن بعلت حلالیت کم در آب مورد توجه میباشند. زیرا با آب فعل و انفعال شیمیائی انجام نداده و حلالیت آنها مستقیماً به فشار جزئی آنها بستگی دارد. حلالیت اکسیژن دردرجه حرارتهای مختلف آب متفاوت است. مقدار حلالیت اکسیژن اتمفسر در آب نسبتاً خالص از 6/14 میلیگرم در لیتر در صفر درجه سانتیگراد تا 7 میلیگرم در لیتر در 35 درجه سانتیگراد تحت فشار 1 اتمسفر متفاوت است. اکسیژن به مقدار کم در آب محلول بوده و حلالیت آن با فشار اتمسفر و درجه حرارت متغیر است. کمبود حلالیت اکسیژن در آب یکی از فاکتورهای اصلی است که ظرفیت تصفیه طبیعی آب را کاهش میدهد. لذا تصفیه فاضلابها قبل از ورود به رودخانهها ضروری میگردد. اکسیژن محلول رودخانهها نیز میتوان آلودگی آنها را کنترل نمود. اکسیژن فاکتور مهمی در ایجاد خورندگی آهن و فولاد بخصوص در سیستمهای توزیع آب و بویلرها میباشد. لذا تعیین مقدار اکسیژن محلول برای کنترل خورندگی آب به کار میرود.
اساس روش اندازهگیری:
معمولیترین روش تعیین اکسیژن محلول بر اساس آزاد کردن ید توسط اکسیژن محلول میباشد. ید آزاد شده عموماً توسط یک محلول احیاءکننده مانند تیوسولفات سدیم اندازهگیری میشود. چسب نشاسته خاتمه عمل را نشان میدهد. نشاسته ید آزاد شده را جذب نموده و رنگ آبی ایجاد میکند و هنگامیکه تمامی ید احیاء گردید محلول بیرنگ میشود.
روش و نیکلر یا ید و متری روش استانداردی برای تعیین اکسیژن محلول میباشد. این روش بر اساس اکسیداسیون Mn2+ به ظرفیت بالاتر (Mn4+) در محیط قلیائی توسط اکسیژن میباشد. منگنز با ظرفیت چهار یون ید را در محیط اسیدی به ید آزاد اکسیده میکند و مقدار ید آزاد شده که برابر با اکسیژن محلول میباشد توسط تیتزاسیون با تیوسولفات استاندارد اندازهگیری میشود.
در این روش وجود بعضی از مواد مانند نیتویتها و آهن سه ظرفیتی قابلیت اکسیدکنندگی I- را به I2 دارند. و نتیجه آزمایش را زیاد نشان میدهد. و بر عکس مواد احیاء کننده مانند S2-, So32-, Fe2+ ید را احیاء کرده و به I- تبدیل میکنند و نتیجه آزمایش را کمتر از حد واقعی نشان میدهد. بنابراین روش تصحیح نشده چنانکه اکسیژن در محیط وجود نداشته باشد با افزایش سولفات منگنز و معرف یدور قلیائی (NaOH, KI) رسوب سفید رنگ 2Mn(OH) تشکیل میگردد.
Mn2+ + 2OH- Mn(OH)2
رسوب سفید رنگ
اگر در محیط اکسیژن موجود باشد مقداری از Mn2+ به ظرفیت بالاتر اکسیده شده و رسوب قهوهای رنگ Mno2 ظاهر میگردد.
Mn(OH)2 + ½ O2 Mno2 + H2O
رسوب قهوهای رنگ
اکسیداسیون Mn2+ به Mno2 به آهستگی انجام میگیرد بدین منظور محلول تکان داده میشود تا تمام اکسیژن محلول به صورت ترکیب درآید. حرکت دادن محلول برای مدت حداقل 20 ثانیه لازم است در صورتیکه آب مورد آزمایش شور باشد. زمان تماس بایستی به مراتب طولانیتر باشد. پس از حرکت دادن، نمونه را برای ترکیب کامل اکسیژن در جائی ساکن گذاشته و پس از مدتی به آن اسید سولفوریک اضافه میکنند. در محیط اسیدی Mno2 یون I- را اکسید کرده و ید آزاد میکند. برای انجام واکنش بطور کامل بایستی درب بطری را بست و برای حداقل 10 ثانیه آن را حرکت داد تا ید به طور یکسان در تمام محلول پخش گردد. ید حاصله را با تیوسولفات سدیم استاندارد تیتر میکنند.
Mno2 + 2 I- + 4H+ Mn2+ + I2 + 2H2O
I2 + 2 Na2S2o3 Na2S4O6 + 2 NaI
همانطور که اشاره شد یون نیتریت یکی از مواد مزاحم در تعیین اکسیژن محلول مبباشد. یون نیتریت Mn2+ را اکسیده نمیکند ولی I- را در محیط اسیدی به I2 اکسید مینماید. لذا اگر نیتریت در محیط وجود داشته باشد تشخیص نقطه پایانی مشکل خواهد بود. زیرا به محض اینکه رنگ آبی معرف نشاسته و ید محو میگردد نیتریتها مقداری از I- را به I2 اکسید کرده و رنگ آبی دوباره ظاهر میگردد. برای حذف دخالت نیتریتها از آز ید سدیم (Nan3) استفاده میشود.
NaN3 + 9H+ 3NH3 + Na+ + NH3 + NO2- + H+ N2 + N2O + H2O
HN3 + NO2- + H+ N2 + N2O + H2O
بدین طریق دخالت نیتریتها میگردد.
معرفهای لازم:
محلول سولفات منگنز: مقدار 480 گرمMnSO4.4H20 یا 400 گرم MnSO4. 2H2O و یا 364 گرم MnSO4.H2O را در آب مقطر حل کرده و حجم آن را به یک لیتر برسانید.
معرف یدور و آز ید قلیائی: مقدار 500 گرم از NaOH را با 150 گرم یدور پتاسیم در آب مقطر حل کرده و حجم آن را به یک لیتر برسانید. به آن مقدار 10 گرم آز ید سدیم که در 40 سی سی آب مقطر حل شده اضافه کنید.
- اسید سولفوریک غلیظ
- نشاسته: 2 گرم نشاسته و 2/0 گرم اسید سالیسیلیک را در 100 سی سی آب مقطر گرم حل کنید.
- سدیم تیوسولفات استاندارد M 025/0: 205/6 گرم از Na2S2O3. 5H2O را در آب مقطر جوشیده سرد شده حل کرده به آن 4/0 گرم سود افزوده و در بالن ژوژه یک لیتری به حجم برسانید. این محلول را در مقابل محلول استاندارد 025/0 نرمال بیکرمات پتاسیم و یا بی یدات پتاسیم استاندارد کنید.
روش کار:
به نمونه مورد آزمایش که در بطریهای 250 سیسی یا 300 سی سی جمعآوری شده مقدار 1 سی سی محلول سولفات منگنز و 1 سی سی معرف یدور و آزید قلیائی اضافه کنید. سپس درب بطری را با دقت بسته چندین بار آن را تکان داده تا رسوب هیدروکسید منگنز ظاهر گردد. بعد از تهنشین شدن رسوب مقدار 1 سی سی اسید سولفوریک غلیظ به آن افزوده و آن را مخلوط کنید تا رسوب بطور کامل حل شود، 200 سی سی از محلول فوق را در یک ارلن ریخته و با تیوسولفات N 025/0 تا رنگ زرد کم رنگ تیتر کنید. سپس چند قطره چسب نشاسته به آن افزوده و تیتراسیون را تا بی رنگ شدن محلول ادامه دهید. حجم تیوسولفات مصرفی را یادداشت کنید. با استفاده از معادلههای واکنش مقدار اکسیژن محلول نمونه را بر حسب میلیگرم در لیتر محاسبه کنید.
Nitrous Oxide و خواص آن
Nitrous oxide, N2O, is a colorless, almost odorless gas, that was first discovered in 1793 by the English scientist and clergyman Joseph Priestley (who was also famous for being the first to isolate other important gases such as oxygen, carbon monoxide, carbon dioxide, ammonia, and sulfur dioxide). Priestley made N2O by heating ammonium nitrate in the presence of iron filings, and then passing the gas that came off (NO) through water to remove toxic by-products. The reaction he observed was:
2NO + H2O + Fe ![]() N2O + Fe(OH)2
N2O + Fe(OH)2
After initial trials, Priestley thought that N2O could be used as a preserving agent, but this proved unsuccessful.
افزایش گاز اکسید نیتروژن دو خطر را همراه دارد: 1- این گاز از گازهای گلخانه ای محسوب میگرددکه طول عمر زیادی در اتمسفر دارد (حدود 120 سال) و تاثیر آن در توانایی ایجاد پدیده گرم شدن زمین 310 برابر گاز دی اکسید کربن است 2- گاز اکسید نیتروژن آزاد شده، عاقبت در قسمت فوقانی جو به گاز نیتریک اکساید تبدیل می گردد که گاز اخیر توانایی شکستن ازن را دارد. با این توصیف افزایش سطح اکسید نیتروژن نه تنها در اثر گلخانه ای سهم عمده دارد، بلکه ممکن است بطور غیر مستقیم در شدت تشعشع پرتوهای ماورای بنقش نیز تاثیر گذارد.
آشنایی با روشهای تفکیک و تخلیص ترکیبات آلی
شیمیدان آلی باتجربه به ندرت واکنش هایی را می یابد که فقط محصول (یا محصولات)مورد نظرش را تولید کند.علت این عمل آن است که در مخلوط واکنش همراه با محصول مورد نظر مقادیر مختلفی از مواد اولیه تغییر نیافته و حلال ومحصول واکنشهای جانبی که همزمان با واکنش اصلی انجام می شوند وجود دارد.
شیمیدان نیروی زیادی را صرف می کند تا محصول مورد نظر را از چنین ناخالصی هایی جدا کند.هدف از این تحقیق ارائه روشهای مهمی است که شیمیدان امروزی جهت تفکیک و تخلیص ترکیبات آلی به کار برد.
تقطیر
روشهای مختلفی برای جداسازی مواد اجزای سازنده یک محلول وجود دارد که یکی از این روشها فرایند تقطیر میباشد در روش تقطیر جداکردن اجزاء یک مخلوط ، از روی اختلاف نقطه جوش آنها انجام میگیردتقطیر ، در واقع ، جداسازی فیزیکی برشهای نفتی است که اساس آن ، اختلاف در نقطه جوش هیدروکربنهای مختلف است. هر چه هیدروکربن سنگینتر باشد، نقطه جوش آن زیاد است و هر چه هیدروکربن سبکتر باشد، زودتر خارج میشود.. تقطیر در عمل به دو روش زیر انجام میگیرد. روش اول شامل تولید بخار از طریق جوشاندن یک مخلوط مایع ، سپس میعان بخار ، بدون اینکه هیچ مایعی مجددا به محفظه تقطیر بازگردد. در نتیجه هیچ مایع برگشتی وجود ندارد. در روش دوم قسمتی از بخار مایع شده به دستگاه تقطیر باز میگردد و به صورتی که این مایع برگشتی در مجاورت بخاری که به طرف مبرد میرود قرار میگیرد. هر کدام از این روشها میتوانند پیوسته یا ناپیوسته باشند.
تقطیر، معمولترین روشی است که برای تخلیص مایعات به کار می رود. دراین عمل مایع را به کمک حرارت تبخیر می کنند و بخار مربوطه را در ظرف جداگانه ای متراکم می کنند و محصول تقطیر را بدست می آورند. چنانچه ناخالصیهای موجود در مایع اولیه فرار نباشند، در باقی مانده تقطیر به جا می مانند و تقطیر ساده جسم را خالص میکند. در صورتی که ناخالصیها فرار باشند، تقطیر جزء به جزء مورد احتیاج خواهد بود.
چنانچه ناخالصی های موجود در مایع اولیه فرار نباشد در باقیمانده تقطیر به جا می ماند و تقطیر ساده نمونه را خالص می کند.در صورتیکه فرار باشند تقطیر جز به جز مورد نیاز خواهد بود.اگر فقط یک ماده فرار بوده و اختلاف نقطه ی جوش این ماده با ناخالصی های موجود در آن زیاد باشد (حدود 30درجه)می توان برای جدا کردن این ماده از ناخالصی ها از تقطیر ساده استفاده نمود.از تقطیر ساده معمولا د جداسازی مخلوط مایعاتی استفاده می شود که نقطه یجوشی در محدوده 40تا150درجه دارندزیرا در دمای بالاتر از 150درجه بسیاری از ترکیبات آلی تجزیه می شوندودر دمای جوش کمتر از 40درجه مقدار زیادی از مایع در ضمن تقطیرهدر می رود.
در تقطیر مخلوطی ازدو یا چند جسم فشاربخار کل تابعی از فشار بخار هر یک از اجزا و کسر مولی آنه می باشد. بر اساس قانون رائول فشار بخار جزیی یک ترکیب فرار در یک محلول ایده آل با حاصلضرب فشار بخار در کسر مولی آن برابر است.بنابراین در بخار موجود بر سطح دو یا چند جزمحلول فرار ذرات کلیه اجزا شرکت کننده در محلول یافت می شود.رابطهی بین فشار بخار کل(Pt)با فشار جزیی (Pi)و کسر مولی اجزا(Xi)به صورت زیر است:
Pt=PaXa+PbXb+PcXc+……
نکته:اگر در محلولی شامل دو ماده شیمیایی فرار یک جز دارای فشار بخار بیشتری از جز دیگر باشد بخار حاصل از آن در مقایسه با مایع دارای درصد بیشتری از جسم فرارتر خواهد بود.
ظروف معمولی در خلل و شکاف های جدار خود دارای بسته ها ی هوای محبوس می باشند.با ریختن مایع در ظرف محفظه بسته ها از بخار پر می شود.وقتی که دمای مایع افزایش می یابد بخار آنقدر به حالت متراکم باقی می ماند تا اینکه از فشار بخار روی مایع بیشتر شود.در این حالت بخار به دام افتاده افزایش حجم پیدا می کند و به صورت حباب هایی به سطح مایع رسیده و خارج می گردد. حالت به هم خوردگی حاصل از حباب ها (جوش)حباب های هوای بیشتری را به داخل مایع کشانده و فرایند با تشکیل بخار ادامه می یابد.
با حرارت دادن مایعات درظروف شیشه ای که دارای سطوحی نسبتا صاف و یکنواخت می باشند حالت جوش ایجاد نمی شود و اگر درجه حرارت به اندازه کافی افزایش یابد به حالت انفجاری تبخیر می گردند.برای اجتناب از خطرات مربوط به جوشش ناگهانی (به صورت ضربه ای)منبعی برای دمیدن حباب ها به درون مایع قبل از حرارت دادن و عمل جوش لازم است. در شرایط معمولی (فشارجو)این منبع سنگ جوش می باشد.سنگ جوش دانه هایی حاوی خلل ریز در خود بوده که در آن مولکولهای هوا حبس شده اند.با قرار گرفتن این دانه ها در حلول حباب ها از سطح آنها تشکیل شده واز جوشیدن انفجاری و تاخیر در جوش جلوگیری می نماید.
در ادامه به معرفی انواع روشهای تقطیر و توضیح اجمالی در ارتباط با آنها پرداخته ایم:
انواع تقطیر :
تقطیر ساده:
به عنوان مثال هنگامیکه ناخالصی غیر فراری مانند شکر به مایع خالصی اضافه می شود فشار بخار مایع تنزل می یابد.علت این عمل آن است که وجود جز غیر فرار به مقدار زیادی غلظت جز اصلی فرار را پایین می آورد یعنی دیگر تمام مولکولهایی که در سطح مایع موجودند مولکولهای جسم فرار نیستند و بدین ترتیب قابلیت تبخیر مایع کم می شود.نمودار ارائه شده در زیر اثر جز غیر فرار را در فشار بخار مخلوط نشان می دهد:
تقطیر ساده را می توان به دوصورت تعریف کرد:1-تقطیر ساده غیر مداوم2-تقطیر ساده مداوم
• تقطیر ساده غیر مداوم : در این روش تقطیر ، مخلوط حرارت داده میشود تا بحال جوش درآید بخارهایی که تشکیل میشود غنی از جزء سبک مخلوط میباشد پس از عبور از کندانسورها (میعان کننده ها) تبدیل به مایع شده ، از سیستم تقطیر خارج میگردد. به تدریج که غلظت جزء سنگین مخلوط در مایع باقی مانده زیاد میشود، نقطه جوش آن بتدریج بالا میرود. به این ترتیب ، هر لحظه از عمل تقطیر ، ترکیب فاز بخار حاصل و مایع باقی مانده تغییر میکند.
• تقطیر ساده مداوم : در این روش ، مخلوط اولیه (خوراک دستگاه) بطور مداوم با مقدار ثابت در واحد زمان ، در گرم کننده گرم میشود تا مقداری از آن بصورت بخار درآید، و به محض ورود در ستون تقطیر ، جزء سبک مخلوط بخار از جزء سنگین جدا می شود و از بالای ستون تقطیر خارج میگردد و بعد از عبور از کندانسورها ، به صورت مایع در میآید جزء سنگین نیز از ته ستون تقطیر خارج میشود. قابل ذکر است که همیشه جزء سبک مقداری جزء سنگین و جزء سنگین نیز دارای مقداری از جزء سبک است.
نکته:در تقطیر یک ماده خالص چنانچه مایع زیاده از حد گرم نشوددرجه حرارتی که در گرماسنج دیده می شود یعنی درجه حرارت دهانه ی خروجی با درجه حرارت مایع جوشان در ظرف تقطیر یعنی درجه حرارت ظرف یکسان است.درجه حرارت دهانه خروجی که به این ترتیب به نقطه جوش مایع مربوط می شود در طول تقطیر ثابت می ماند.
هرگاه در مایعی تقطیر می شود ناخالصی غیر فراری موجود باشد درجه حرارت دهانه خروجی همان درجه حرارت مایع خالص است زیرا ماده ای که بر روی حباب گرماسنج متراکم می شود به ناخالصی آلوده نیست.ولی درجه حرارت ظرف به علت کاهش فشا بخار محلول بالا می ررود. در جریان تقطیر درجه حرارت ظرف نیز افزایش می یابد.زیرا که غلظت ناخالصی با تقطیر جز فرار به تدریج زیاد می شود و فشار بخار مایع بیشتر پایین می اید.با وجود این درجه حرارت دهانه خروجی مانند مایع خالص ثابت می ماند.رابطه کمی موجود بین فشار بخاروترکیب مخلوط همگن مایع(محلول)به قانون رائول معروف است وبه صورت معادله زیربیان می شود:
جز مولی Rبه جزیی اطلاق می شود که تمام مولکولهای موجود در آن مولکولهای Rباشند.برای به دست آوردن این جز مولی تعداد مولهای Rدر مخلوط را بر مجموع تعداد مولهای اجزا سازنده تقسیم می کنند.معادله در زیر آمده است:
باید دانست که در بالای محلول ایده آلی که محتوی Rاست فشار بخار جزR فقط به جزمولی Rبستگی داردوبه هیچ وجه به فشار بخار اجزای دیگر مربوط نیست.چنانچه کلیه اجزا به غیر از Rغیر فرار باشند فشار بخار کلی مخلوط برابر با فشار جز Rاست زیرا می توان فشار بخار ترکیبات غیر فرار را صفر فرض کرد.در نتیجه محصول تقطیر چنین مخلوطی همیشه Rخالص است.ولی اگر دو یا چند جز فرار باشند در این صورت فشار بخار کل برابر با مجموع فشار بخارهای جزیی هر یک از اجزای فرار خواهد شد.(قانون دالتون-در اینجا RوSوTفقط به اجزای فرار مربوط می شود):
چنین مخلوط مایعی که در بالا توضیح داده شد تفاوت زیادی دارد زیرا در اینجا ممکن است محصول تقطیر هر یک از اجزای فراررا در بر داشته باشد.تفکیک دراین حالت احتیاج به تقطیر جز به جز دارد.چگونگی انجام تقطیر جز به جز در ادامه آمده است.
• تقطیر تبخیر آنی (ناگهانی): وقتی محلول چند جزئی مانند نفت خام را حرارت میدهیم، اجزای تشکیل دهنده آن بترتیب که سبکتر هستند، زودتر بخار میشود. برعکس وقتی بخواهیم این بخارها را سرد و دوباره تبدیل به مایع کنیم، هر کدام که سبکتر باشد دیرتر مایع میگردد. با توجه به این خاصیت ، میتوانیم نفت خام را به روش دیگری که به آن "تقطیر آنی" گویند، تقطیر نماییم. در این روش ، نفت خام را چنان حرارت میدهیم که ناگهان همه اجزای آن تبدیل به بخار گردد و سپس آنها را سرد میکنیم تا مایع شود. در اینجا ، بخارها به ترتیب سنگینی ، مایع میشوند یعنی هرچه سنگینتر باشند، زودتر مایع میگردند و بدین گونه ، اجزای نفت خام را با ترتیب مایع شدن از هم جدا میکنیم.
• تقطیر در خلا : با توجه به اینکه نقطه جوش مواد سنگین نفتی نسبتا بالاست و نیاز به دما و انرژی بیشتری دارد، و از طرف دیگر ، مقاومت این مواد در مقابل حرارت بالا کمتر میباشد و زودتر تجزیه میگردند، لذا برای جداکردن آنها از خلا نسبی استفاده میشود. در این صورت مواد دمای پایینتر از نقطه جوش معمولی خود به جوش میآیند. در نتیجه ، تقطیر در خلا ، دو فایده دارد: اول این که به انرژی و دمای کمتر نیاز است، دوم اینکه مولکولها تجزیه نمیشوند. امروزه در بیشتر موارد در عمل تقطیر ، از خلا استفاده میشود. یعنی این که: هم تقطیر جزء به جزء و هم تقطیر آنی را در خلا انجام میدهند.
• تقطیر به کمک بخار آب : یکی دیگر از طرق تقطیر آن است که بخار آب را در دستگاه تقطیر وارد میکنند در این صورت بی آنکه خلاءای ایجاد گردد، اجزای نفت خام در درجه حرارت کمتری تبخیر میشوند. این مورد معمولا در زمانی انجام میشود که در نقطه جوش آب ، فشار بخار اجزای جدا شونده بالا باشد تا به همراه بخار آب از مخلوط جدا گردند.
غالبابه کمک تقطیر با بخار آب می توان ترکیبات آلی فراری را که باآب مخلوط نمی شوند یا تقریبا با آن غیر قابل اختلاط هستند تفکیک و تخلیص کرد.در این روش مخلوط آب وجسم آلی با هم تقطیر می شوند.عمل تقطیر یکمخلوط غیر قابل امتزاج در صورتی که یکی از اجزا آب باشد تقطیر با بخار آب نامیده می شود.
با توجه به اصولی که در تقطیر با بخار آب وجود داردمی توان محاسن ومحدودیت های این روش را به بهترین وجه تشریح کرد.در مخلوطی از مواد فرار و غیر قابل اختلاط فشار جزییpiهر جز در یک درجه حرارت معین برابر با فشار بخار piترکیب خالص در همان درجه حرارت استو به جز مولی ترکیب در مخلوط بستگی نداردیعنی هر یک از اجزای سازنده مخلوط به طور مستقل از اجزای دیگر تبخیر می شوند.
این حالت با مخلوط مایعات قابل اختلاط اختلاف زیادی دارد زیرا که در این مایعات فشار جزیی هر جز سازنده به جز مولی آن در محلول مربوط است.(قانون رائول)در مخلوط ترکیبات فرار وغیر قابل اختلاط بر طبق قانون دالتون فشار کلی Ptمحلول (مخلوط)گازها با مجموع فشارهای جزیی گازهای تشکیل دهنده می شودو به این ترتیب فشار بخار کلی این مخلوط از معادله زیر به دست می آید:
از این عبارت چنین ر می آید که همواره در هر درجه حرارتی فشار بخار کل مخلوط حتی از فشار بخار فرارترین جز در آن درجه حرارت بیشتر است زیرا که فشار بخار اجزای دیگر مخلوط هم دخالت می کنند.بنابراین باید درجه جوش مخلوط ترکیبهای غیر قابل اختلاط کمتر از جزیی باشد که کمترین نقطه جوش را دارد.درجه حرارت تقطیر با بخار آب نسبتا پایین (100درجه یا کمتراز آن)است و این تقطیر به خصوص در تخلیص موادی به کار می رود که نسبت به حرارت حساسیت دارندودر حرارت های بالا تجزیه می شوند.هم چنین این روش برای جدا کردن ترکیب از مخلوط از مخلوط واکنشی که محتوی مقدار زیادی از مواد (قیرمانند)باشد مفید است.این مواد غیر فرار و بی مصرف در اغلب واکنشهای آلی تشکیل می شوند.ترکیب درصد محصولی که در تقطیر با بخار آب به دست می آید به وزن مولکولی ترکیبات مورد تقطیر و هم چنین به فشار بخار آنها در درجه حرارت تقطیر مخلوط بستگی دارد.مخلوطی از دو جز غیر قابل اختلاط AوBرا در نظر بگیرید.چنانچه بخارهای AوBمانند گازهای ایده آل عمل می کنند با استفاده از قانون گازهای ایه آل می توان دو عبارت زیر را به دست آورد:
از تقسیم معادله اول به دوم چنین به دست می آید:
چون فاکتور RTدر صورت و مخرج کسر مساوی است و حجم اشغالی گاز برای هر دو یکسان است(VA=VB)عبارت بالا چنین می شود:
فرایند تقطیر با بخار آب در آزمایشگاه و صنعت به طور وسیعی مورد استفاده قرار می گیرد .به عنوان مثال برای جداسازی الفاپی نن-آنیلین-نیتروبنزنوبسیاری از اسانس های طبیعی وروغن های معطر به کار می رود.به طور خلاصه تقطیر با بخار آب روشی را فراهم می کند که به کمک آن می توان ترکیبات آلی مایع و جامدی را که فرار هستند ودر آب حل نمی شوند (یا تقریبا در آن نا محلولند)در شرایط نسبتا ملایم از ترکیبات غیر فرار جدا کرد.مسلما این روش برای موادی که در اثر تماس زیاد با آب گرم تجزیه می شوند یا با اب واکنشی می دهند یا در 100درجه فشار بخارشان 5میلی متر یا کمتر باشد مناسب نیست.
• تقطیر آزئوتروپی : از این روش تقطیر معمولا در مواردی که نقطه جوش اجزاء مخلوط بهم نزدیک باشند استفاده میشود، جداسازی مخلوط اولیه ، با افزایش یک حلال خاص که با یکی از اجزای کلیدی ، آزئوتوپ تشکیل میدهد امکانپذیر است. آزئوتروپ محصول تقطیر یا ته مانده را از ستون تشکیل میدهد و بعد حلال و جزء کلیدی را از هم جدا میکند. اغلب ، ماده افزوده شده آزئوتروپی با نقطه جوش پایین تشکیل میدهد که به آن شکننده آزئوتروپ میگویند. آزئوتروپ اغلب شامل اجزای خوراک است، اما نسبت اجزای کلیدی به سایر اجزای خوراک خیلی متفاوت بوده و بیشتر است.
مثالی از تقطیر آزئوتروپی استفاده از بنزن برای جداسازی کامل اتانول از آب است، که آزئوتروپی با نقطه جوش پایین با 6/95% وزنی الکل را تشکیل میدهد. مخلوط آب- الکل با 95% وزنی الکل به ستون تقطیر آزئوتروپی افزوده میشود و جریان جریان غنی از بنزن از قسمت فوقانی وارد میشود. محصول ته مانده الکل تقریبا خالص است وبخار بالایی یک آزئوتروپی سهگانه است. این بخار مایع شده، به دو فاز تقسیم میشود. لایه آلی برگشت داده شده، لایه آلی به ستون بازیافت بنزن فرستاده میشود. همه بنزن و مقدار الکل در بخار بالایی گرفته شده، به ستون اول روانه میشوند. جریان انتهایی در ستون سوم تقطیر میشود تا آب خالص و مقداری آزئوتروپ دوگانه از آن بدست آید.
• تقطیر استخراجی : جداسازی اجزای با نقطه جوش تقریبا یکسان از طریق تقطیر ساده مشکل است حتی اگر مخلوط ایده آل باشد و به دلیل تشکیل آزئوتروپ ، جداسازی کامل آنها غیر ممکن است برای چنین سیستم هایی با افزایش یک جزء سوم به مخلوط که باعث تغییر فراریت نسبی ترکیبات اولیه میشود، جداسازی ممکن میشود. جزء افزوده شده باید مایعی با نقطه جوش بالا باشد، قابلیت حل شدن در هر دو جزء کلیدی را داشته باشد و از لحاظ شیمیایی به یکی از آنها شبیه باشد. جزء کلیدی که به حلال بیشتر شبیه است ضریب فعالیت پایین تری از جزء دیگر محلول دارد، در نتیجه جداسازی بهبود می یابد این فرآیند ، تقطیر استخراجی نام دارد.
مثالی از تقطیر استخراجی، استفاده از فور فورال در جداسازی بوتادیان و بوتن است، فورفورال که حلالی به شدت قطبی است، فعالیت بوتادی ان را بیش تر از بوتن و بوتان کم میکند و غلظت بوتادی ان وفورفورال وارد قسمت فوقانی ستون تقطیر استخراجی شود، با انجام تقطیر بوتادی ان از فورفورال جدا میشود.
• تقطیر جزء به جزء : اجزای سازنده محلول شامل دو یاچند فرار را که از قانون رائول پیروی میکنند، میتوان با فرایند تقطیر جزء به جزء از هم جدا کرد. طبق قانون رائول ، فشار بخار محلول برابر با مجموع اجزای سازنده آن است و سهم هر جزء برابر با حاصلضرب کسر مولی آن جزء به جزء در فشار بخار آن در حالت خاص است. در تقطیر محلولی از B و A ، غلظت A در بخاری که خارج شده و مایع میشود، بیش از غلظت آن در مایع باقی مانده است. با ادامه عمل تقطیر ، ترکیب درصد اجزا در بخار و مایع دائما تغییر میکند و این در هر نقطه عمومیت دارد. با جمع آوری مایعی که از سردشدن بخار حاصل میشود و از تقطیر مجدد آن و با تکراری پی در پی این عمل ، سرانجام میتوان اجزای سازنده مخلوط اصلی را به صورتی واقعا خالص بدست آورد.
از نظر سهولت در اینجا فقط محلولهای ایده آل دو تایی را که محتوی دو جز فرار RوSباشند در نظر می گیریم.محلول ایده ال به محلولی اطلاق می شود که در آن اثرات بین مولکولهای متجانس مشابه با اثرات بین مولکولهای غیر متجانس باشد.گرچه فقط محلولهای ایده ال به طور کامل از قانون رائول پیروی می کنند ولی بسیاری از محلولهای آلی به محلولهلی ایده آل نزدیک هستند.
تقطیر جزبه جز محلول های غیر ایده ال
گرچه بیشتر مخلوط های یکنواخت مایع به صورت محلولهای ایده ال عمل می کنندولی نمونه های بسیاری وجود دارد که نحوه عمل آنها ایده آل نیست.در این محلولها مولکولهای غیر متجانس در مجاورت یکدیگر به طور یکسان عمل نمی کنند انحراف حاصل از قانون رائول به دو روش انجام میگیرد:
بعضی از محلولها فشار بخار بیشتری از فشار بخار پیش بینی شده ظاهر می سازندوگفته می شود که انحراف مثبت دارند. بعضی دیگر فشار بخار کمتری از فشار پیش بینی شده آشکار می کنندومی گویند که انحراف منفی نشان می دهند.
در انحراف مثبت نیروی جاذبه بین مولکولهای مختلف دو جز سازنده ضعیف تر از نیروی جاذبه بین مولکولهای مشابه یک جز است و در نتیجه در حدود ترکیب درصد معینی فشار بخار مشترک دو جز بزرگتر از فشار بخار جز خالصی می شود که فرارتر است.بنابراین مخلوط هایی که ترکیب درصد آنها در این حدود باشد درجه جوش کمتری از هر یک از دو جز خالص دارند.مخلوطی که در این حدود حداقل درجه جوشش را دارد باید به صورت جز سوم در نظر گرفته شود.این مخلوط نقطه جوش ثابتی دارد زیرا ترکیب درصد بخاری که در تعادل با مایع است با ترکیب درصد خود مایع برابر است.چنین مخلوطی را آزئوتروپ یا مخلوط آزئوتروپ با جوشش مینی مم می نامند.از تقطیر جز به جز این مخلوط ها هر دو جز به حالت خالص به دست نمی آید بلکه جزیی که ترکیب درصد آن از ترکیب درصد آزئوتروپ بیشتر باشد تولید می شود.
در انحراف منفی از قانون رائول نیروی جاذبه بین مولکولهای مختلف دو جز قویتر از نیروی جاذبه بین مولکولهای مشابه یک جز است ودر نتیجه ترکیب درصد معینی فشار بخار مشترک دو جز کمتر از فشار بخار جز خالص می شودکه فرارتر است.بنابراین مخلوط هایی که ترکیب درصد آنها در این حدود باشد حتی نسبت به جز خالصی که نقطه جوش بیشتری دارد در درجه حرارت بالاتری می جوشند.در اینجا ترکیب درصد به خصوصی وجود دارد که به آزئو تروپ با جوشش ماکسیمم مربوط می شود.تقطیر جز به جز محلولهایی که ترکیب درصدی غیر از ترکیب درصد آزئوتروپ دارندباعث خروج جزیی مخلوط می شودکه ترکیب درصد آن از آزئوتروپ بیشتر باشد.
ستونهای تقطیرجز به جز:
این ستونها انواع متعددی داردولی در تمام آنها خصلت های مشابهی وجود دارد.این ستونها مسیر عمودی را به وجود می آورند که باید بخار در انتقال از ظرف تقطیر به مبرد از آن بگذرد.این مسیر به مقدار قابل ملاحظه ای از مسیر دستگاه تقطیر ساده طویل تر است.هنگام انتقال بخار از ظرف تقطیر به بالای ستون مقداری از بخار متراکم می شود.چنان چه قسمت پایین این ستون نسبت به قسمت بالای آن در درجه حرارت بیشتری نگه داری شود مایع متراکم شده و در حالی که به پایین ستون می ریزد دوباره به طور جزیی تبخیر می شود .بخار متراکم نشده همراه بخاری که از تبخیر مجدد مایع متراکم شدهحاصل می شود در داخل ستون بالاتر می رود واز یک سری تراکم وتبخیر می گذرد.این اعمال باعث تقطیر مجدد مایع می شود و به طوریکه در هر یک از مراحل فاز بخاری که به وجود می آید نسبت به جز فرارتر غنی تر می شود.ماده متراکم شده ای که به پایین ستون می ریزددر مقایسه با بخاری که با آن در تماس است در هر یک از مراحل نسبت جزیی که فراریت کمتری دارد غنی تر می شود.
در شرایط ایده ال بین فازهای مایع و بخار در سراسر ستون تعادل برقرار می شود و فاز بخار بالایی تقریبا به طور کامل از جز فرارتر تشکیل می شود و فاز مایع پایینی نسبت به جزیی که فراریت کمتری دارد غنی تر می شود.
مهم ترین شرایطی که برای ایجاد این حالت لازم است عبارتند از :
1-تماس کامل و مداوم بین فازهای بخار و مایع در ستون 2-حفظ افت مناسبی از درجه حرارت در طول ستون 3-طول کافی ستون 4-اختلاف کافی در نقاط جوش اجزای مخلوط مایع.
چنان چه دو شرط اول کاملا مراعات شود می توان با یک ستون طویل ترکیباتی که اختلاف کمی در نقطه ی جوش دارند به طور رضایت بخش از هم جدا کرد .زیرا طول ستون مورد لزوم و اختلاف نقاط جوش اجزا با هم نسبت عکس دارند.معمول ترین راه ایجاد تماس لازم در بین فازهای مایع آن است که ستون با مقدارری ماده بی اثر مانند شیشه یا سرامیک یا تکه های فلزی به اشکال مختلف که سطح تماس وسیعی را فراهم می کندپر شود. یکی از راه های بسیار موثر ایجاد این تماس بین مایع و بخار آن است که نوار چرخانی از فلز یا تفلون که با سرعت زیاذی در داخل ستون بچرخد به کار رود.
این عمل نسبت به ستون های پر شده ای که قدرت مشابهی دارند این مزیت را دارد که ماده کمی را در داخل ستون نگاه می دارد(منظور از این نگه داری مقدار مایع و بخاری است که برای حفظ شرایط تعادل در داخل ستون لازم است.)
تقطیر تبخیر ناگهانی
در این نوع تقطیر ، مخلوطی از مواد نفتی که قبلا در مبدلهای حرارتی و یا کوره گرم شدهاند، بطور مداوم به ظرف تقطیر وارد میشوند و تحت شرایط ثابت ، مقداری از آنها به صورت ناگهانی تبخیر میشوند. بخارات حاصله بعد از میعان و مایع باقیمانده در پایین برج بعد از سرد شدن به صورت محصولات تقطیر جمع آوری میشوند. در این نوع تقطیر ، خلوص محصولات چندان زیاد نیست.
تقطیر با مایع برگشتی (تقطیر همراه با تصفیه(
در این روش تقطیر ، قسمتی از بخارات حاصله در بالای برج ، بعد از میعان به صورت محصول خارج شده و قسمت زیادی به داخل برج برگردانده میشود. این مایع به مایع برگشتی موسوم است. مایع برگشتی با بخارات در حال صعود در تماس قرار داده میشود تا انتقال ماده و انتقال حرارت ، صورت گیرد. از آنجا که مایعات در داخل برج در نقطه جوش خود هستند، لذا در هر تماس مقداری از بخار ، تبدیل به مایع و قسمتی از مایع نیز تبدیل به بخار میشود.
نتیجه نهایی مجوعه این تماسها ، بخاری اشباع از هیدروکربنهای با نقطه جوش کم و مایعی اشباع از مواد نفتی با نقطه جوش زیاد میباشد.در تقطیر با مایع برگشتی با استفاده از تماس بخار و مایع ، میتوان محصولات مورد نیاز را با هر درجه خلوص تولید کرد، مشروط بر اینکه به مقدار کافی مایع برگشتی و سینی در برج موجود باشد. بوسیله مایع برگشتی یا تعداد سینیهای داخل برج میتوانیم درجه خلوص را تغییر دهیم. لازم به توضیح است که ازدیاد مقدار مایع برگشتی باعث افزایش میزان سوخت خواهد شد. چون تمام مایع برگشتی باید دوباره به صورت بخار تبدیل شود.
امروزه به علت گرانی سوخت ، سعی میشود برای بدست آوردن خلوص بیشتر محصولات ، به جای ازدیاد مایع برگشتی از سینیهای بیشتری در برجهای تقطیر استفاده شود. زیاد شدن مایع برگشتی موجب زیاد شدن انرژی میشود. برای همین ، تعداد سینیها را افزایش میدهند. در ابتدا مایع برگشتی را 100درصد انتخاب کرده و بعد مرتبا این درصد را کم میکنند و به صورت محصول خارج میکنند تا به این ترتیب دستگاه تنظیم شود.
انواع مایع برگشتی
• مایع برگشتی سرد: این نوع مایع برگشتی با درجه حرارتی کمتر از دمای بالای برج تقطیر برگردانده میشود. مقدار گرمای گرفته شده ، برابر با مجموع گرمای نهان و گرمای مخصوص مورد نیاز برای رساندن دمای مایع به دمای بالای برج است.
• مایع برگشتی گرم: مایع برگشتی گرم با درجه حرارتی برابر با دمای بخارات خروجی برج مورد استفاده قرار میگیرد.
• مایع برگشتی داخلی: مجموع تمام مایعهای برگشتی داخل برج را که از سینیهای بالا تا پایین در حرکت است، مایع برگشتی داخلی گویند. مایع برگشتی داخلی و گرم فقط قادر به جذب گرمای نهان میباشد. چون اصولا طبق تعریف اختلاف دمایی بین بخارات و مایعات در حال تماس وجود ندارد.
• مایع برگشت دورانی: این نوع مایع برگشتی ، تبخیر نمیشود. بلکه فقط گرمای مخصوص معادل با اختلاف دمای حاصل از دوران خود را از برج خارج میکند. این مایع برگشتی با دمای زیاد از برج خارج شده و بعد از سرد شدن با درجه حرارتی کمتر به برج برمیگردد. معمولا این نوع مایع برگشتی درقسمتهای میانی یا درونی برج بکار گرفته میشود و مایع برگشتی جانبی هم خوانده میشود. اثر عمده این روش ، تقلیل حجم بخارات موجود در برج است.
نسبت مایع برگشتی
نسبت حجم مایع برگشتی به داخلی و محصول بالایی برج را نسبت مایع برگشتی گویند. از آنجا که محاسبه مایع برگشتی داخلی نیاز به محاسبات دقیق دارد، لذا در پالایشگاهها ، عملا نسبت مایع برگشتی بالای برج به محصول بالایی را به عنوان نسبت مایع برگشتی بکار میبرند.
تقطیر نوبتی
این نوع تقطیرها در قدیم بسیار متداول بوده، ولی امروزه بعلت نیاز نیروی انسانی و ضرورت ظرفیت زیاد ، این روش کمتر مورد توجه قرار میگیرد. امروزه تقطیر نوبتی ، صرفا در صنایع دارویی و رنگ و مواد آرایشی و موارد مشابه بکار برده میشود و در صنایع پالایش نفت در موارد محدودی مورد استفاده قرار میگیرد. بنابراین در موارد زیر ، تقطیر نوبتی از نظر اقتصادی قابل توجه میباشد.
• تقطیر در مقیاس کم
• ضرورت تغییرات زیاد در شرایط خوراک و محصولات مورد نیاز
• استفاده نامنظم از دستگاه
• تفکیک چند محصولی
• عملیات تولید متوالی با فرآیندهای مختلف
تقطیر مداوم
امروزه بعلت اقتصادی بودن مداوم در تمام عملیات پالایش نفت از این روش استفاده میشود. در تقطیر مداوم برای یک نوع خوراک مشخص و برشهای تعیین شده شرایط عملیاتی ثابت بکار گرفته میشود. بعلت ثابت بودن شرایط عملیاتی در مقایسه با تقطیر نوبتی به مراقبت و نیروی انسانی کمتری احتیاج است. با استفاده از تقطیر مداوم در پالایشگاهها مواد زیر تولید میشود:
گاز اتان و متان بعنوان سوخت پالایشگاه ، گاز پروپان و بوتان بعنوان گاز مایع و خوراک واحدهای پتروشیمی ، بنزین موتور و نفتهای سنگین بعنوان خوراک واحدهای تبدیل کاتالیستی برای تهیه بنزین با درجه آروماتیسیته بالاتر ، حلالها ، نفت سفید ، سوخت جت سبک و سنگین ، نفت گاز ، خوراک واحدهای هیدروکراکینگ و واحدهای روغن سازی ، نفت کوره و انواع آسفالتها.
تقطیر ساده:
شکل دستگاه تقطیر ساده:
1- شعله 2- بالن ته گرد 3- سه راهی تقطیر 4- دماسنج 5- سرد کننده 6- ورودی آب 7- خروجی آب 8- بالن 9- خروج هوا وبخار 10- رابط خلاء
بخش عملی
الف) تقطیر ساده تتراکلریدکربن
20 میلی لیتر تتراکلرید کربن را در بالن تقطیر 50 میلی لیتری بریزید (احتیاط: هرگز از بالنی که بیش از نصف آن از ماده پر شده است استفاده نکنید) دستگاه تقطیر ساده را مطابق شکل سوار کنید و توجه نمائید که حباب دماسنج یا مخزن جیوه ای درست زیر بازوی جانبی بالن تقطیر (محل خروج بخار از بالن) باشد. قطعه کوچکی از سنگ جوش اضافه کنید تا امکان تاخیر در جوش که سبب میشود مایع ناگهانی بالا آید و یا به طور غیر منتظره بداخل مبرد پرت شود، از بین برود.
بالن را با شعله کم حرارت دهید و طوری شعله را تنظیم کنید که سرعت ریختن مایع حاصل از سرد شدن که از مبرد به داخل ظرف جمع آوری می ریزد حدود یک قطره در ثانیه باشد. نموداری از تغییرات درجه حرارت نسبت به حجم مایع جمع آوری شده رسم نمائید و درجه حرارتی که مایع بیشتری تقطیر میشود به عنوان نقطه جوش یاداشت نمائید. تقطیر را در حالی که 3-2 میلی لیتر مایع در بالن تقطیر مانده است قطع کنید.
نقطه جوشی که به دست آورده اید با نقطه جوش کربن تترا کلرید که در کتاب یا مقالات ذکر شده است مقایسه نمائید.
ب) تقطیر ساده متانول و آب
در یک بالن 100 میلی لیتری مخلوطی از 25 میلی لیتر متانول و 25 میلی لیتر آب بریزید. دو عدد سنگ جوش کوچک در بالن بیندازید و به آرامی بالن را حرارت دهید. درجه حرارتی که اولین قطره مایع از نوک ترمومتر به داخل بالن میچکد (میعان) یادداشت کنید و به عنوان شروع تقطیر در نظر بگیرید. در همین لحظه بخارات داخل لوله جانبی شده و مایع میشود و سرازیر شده از دهانه خروجی مبرد وارد ظرف جمع آوری میشود. در ابتدای شروع تقطیر حرارت را به گونه ای تنظیم کنید که سرعت تقطیر یک قطره در ثانیه باشد. دمای ترمومتر را بر حسب حجم تقطیر شده یادداشت کنید و منحنی آنرا رسم کنید.
در فشار 760 mmHg متانول در 7/64 درجه سانتیگراد و آب در oC 100 می جوشد. توجه داشته باشید که در فشار آزمایشگاه در دمای پایین تری تقطیر متانول شروع خواهد شد. هنگامی که 3-2 میلی لیتر مایع در ته بالن باقی مانده است تقطیررا متوقف کنید.
تقطیر جزء به جزء:
برای جداکردن موادی که نقطه جوش آنها خیلی به هم نزدیک باشد از تقطیر جزء به جزء استفاده میکنند. اختلاف این روش با تقطیر ساده آن است که در این حالت از یک ستون تقطیر جزء به جزء استفاده میشود.
ستونهای تقطیر جزء به جزء انواع متعددی دارند ولی در تمام آنها چند خصلت کلی مشاهده میشود. این ستونها مسیر عمودی را به وجود می آورند که باید بخار در انتقال از ظرف تقطیر به مبرد از آن بگذرد، این مسیر به مقدار قابل ملاحظه ای از مسیر دستگاه تقطیر ساده طویلتر است. هنگام انتقال بخار از ظرف تقطیر به بالای ستون مقداری از بخار متراکم میشود. مایع متراکم شده، در حالی که به پایین ستون می ریزد دوباره در تماس با بخاری که از پایین به بالا در جریان است به طور جزئی تبخیر میشود و به سمت بالا میرود و طی این میعان و تبخیر شدنهای متوالی بخار از جزء فرار تر غنی تر میشود، یعنی هرچه به سمت بالای ستون پیش میرویم غلظت جزء فرار تر بیشتر و هر چه به سمت پایین می آییم غلظت جزء غیر فرار بیشتر میشود.
از نقطه نظر تئوری، جدا کردن دو ترکیب فرار به طور کامل، بوسیله تقطیر حتی زمانیکه اختلاف در نقطه جوش آنها زیاد باشد امکان پذیر نیست زیرا همیشه جزء دارای نقطه جوش پایین تر فشار بخارش را بر روی نقطه جوش جزء دیگر اعمال نموده و پاره ای از مولکولهای با نقطه جوش بالاتر نیز تقطیر میگردند. اما بهرحال در امور تجربی، بوسیله تقطیر جزء به جزء میتوان مخلوط اینگونه مایعات را در حد مطلوبی جدا نمود.
تقطیر جزء به جزء مخلوطهای دو جزئی و چند جزئی
هدف از تقطیر ، جداسازی خوراک به بخارهایی از محصولات تقریبا خالص است در تقطیر سیستم های دو جزئی ، درجه خلوص با کسر مولی جزء سبک در محصول تقطیر XO و در محصول ته مانده XB بیان میشود. در سیستم های دو جزئی از یک مرحله به مرحله دیگر ، به جزء در نقطه آزئوتروپ ، دما و منحنی تعادل تغییر میکنند و یک جزء در تمام ستون فرارتر است. اما در سیستم های چند جزئی یک جزء ممکن است در یک قسمت ستون فرارتر و در قسمت دیگر فراریت کمتری داشته باشد، که ماهیت پیچیده غلظت اجزا را نشان میدهد. تعادل فازی سیستم های چند جزئی نسبت به دو جزئی بسیار پیچیده است، به دلیل اینکه تعداد اجزاء زیاد است وتعادل به دما بستگی دارد و دما از یک مرحله به مرحله دیگر تغییر میکند.
شکل دستگاه تقطیر جزء به جزء:
1- سنگ جوش 2- مخلوط دو یا چند ماده 3- گرم کننده 4- ظرف تقطیر (بالن) 5- ستون تقطیر 6- دماسنج 7- خروجی آب 8- ورودی آب 9- سرد کننده 10- رابط خمیده ساده 11- ظرف گیرنده (استوانه مدرج) 12- محصول تقطیر
مخلوط دو ماده با هم در برخی مواد تولید آزئوتروپ میکند، یعنی مخلوط با درصد معینی تا آخرین قطره تقطیر میشود. در اینگونه موارد نمیتوان مخلوط را بوسیله تقطیر جزء به جزء از یکدیگر جدا کرد. برای از بین بردن این حالت یا ماده دیگری به مخلوط اضافه میکنند تا آزئوتروپ دیگری که مطلوب باشد بدست آید و یا فشار را تغییر میدهند. مثلا الکل 95 درصد تشکیل آزئوتروپ میدهد که برای از بین بردن نقطه آزئوتروپ، بنزن به آن اضافه میکنند که در نتیجه نقطه آزئوتروپ دیگری با درصد آب بیشتر ایجاد میشود که بدین ترتیب آب خارج شده، الکل و بنزن باقی میماند که بوسیله تقطیر جزء به جزء به راحتی جدا میشود
بخش عملی
الف)تقطیر جزء به جزء متانول و آب
در یک بالن ته گرد 100 میلی لیتری مقدار 30 میلی لیتر متانول و 30 میلی لیتر آب بریزید و برای اطمینان از جوشش آرام (جلوگیری از غلیان محلول)، چند عدد سنگ جوش اضافه کنید دستگاه تقطیر جزء به جزء را مطابق شکل سوار کنید. از ابتدای شروع تقطیر حرارت را به گونه ای تنظیم کنید که سرعت تقطیر 10 الی 20 قطره در دقیقه باشد. درجه حرارتی که اولین قطره مایع از نوک دماسنج میچکد را یادداشت کنید. اگر ستون مایع طغیان میکند سرعت تقطیر را کم کنید. محصول تقطیر (مقطره) را در سه ظرف جدا در محدوده دمایی زیر جمع آوری نمایید.
تا دمای 68 درجه مقطره را در ظرف (الف) ذخیره کنید.
از 68 درجه تا 90 درجه مقطره را در ظرف (ب) جمع آوری نمایید.
از 90 درجه به بعد، آنرا در ظرف (ج) ذخیره کنید.
تقطیر را ادامه دهید تا 3-2 میلی لیتر مایع در ظرف تقطیر باقی بماند و سپس شعله را خاموش کنید.
حجم مایعات جمع آوری شده در هر ظرف را اندازه گیری کرده و یاد داشت کنید. حجم مایع باقی مانده در ظرف تقطیر را نیز اندازه گیری نموده و یادداشت کنید.
ب) تقطیر جزء به جزء بنزن و تولوئن
در یک ظرف ته گرد 100 میلی لیتری 30 میلی لیتر بنزن و 30 میلی لیتر تولوئن ریخته و برای اطمینان از جوشش آرام، چند عدد سنگ جوش به آن اضافه کنید. دستگاه تقطیر جزء به جزء را آماده کنید.
در این دستگاه محل حباب دماسنج اهمیت ویژه ای دارد، به محل آن نسبت به لوله جانبی سر دستگاه تقطیر توجه کنید (شکل دستگاه تقطیر). سه ظرف 50 میلی لیتری به عنوان ظرف گیرنده با برچسب (الف)، (ب) و (ج) آماده کنید. در عمل باید نوک رابط خلأ تا داخل گردن این ظرف امتداد داشته باشد، بین رابط و ظرف گیرنده یک فضای عمودی باقی نگذارید زیرا این فضا باعث سهولت فرار بخارهای قابل اشتعال میشود.
ظرف تقطیر را با چراغ گاز حرارت دهید. چراغ را طوری قرار دهید که نوک شعله با توری سیمی تماس پیدا کند یا درست زیر آن باشد، و شعله را از جریان باد محفوظ نگه دارید به نحوی که بتوانید حرارت را تا حد ممکن به دقت تنظیم کنید. به مجردی که محلول شروع به جوشیدن کرد و بخارهای رفلاکس شده به گرما سنج رسید، شعله را طوری میزان کنید که تقطیر فقط با سرعتی در حدود یک قطره مایع مقطر در هر یک یا دو ثانیه به طور یکنواخت ادامه یابد. اولین مایع مقطر را در ظرف گیرنده (الف) جمع آوری کنید. وقتی که درجه حرارت دهانه خروجی به 80 درجه رسید، ظرف گیرنده (الف) را با ظرف گیرنده (ب) و در 105 درجه آن را با ظرف گیرنده (ج) عوض کنید. تقطیر را ادامه دهید تا حدود 2 میلی لیتر مایع در ظرف تقطیر باقی بماند و بعد شعله را خاموش کنید. حجم اجزاء تقطیر شده در ظرف گیرنده (الف)، (ب) و (ج) را به کمک استوانه مدرج اندازه بگیرید و یاداشت کنید. اجازه دهید تا مایع موجود در ستون تقطیر به داخل ظرف تقطیر برگردد، حجم باقی مانده را اندازه گرفته و یادداشت کنید.
تقطیر با بخار آب:
غالبا به کمک تقطیر با بخار آب میتوان ترکیبات آلی فراری را که با آب مخلوط نمیشوند یا تقریبا با آن غیر قابل اختلاط هستند تفکیک و تخلیص کرد. در این روش مخلوط آب و جسم آلی با هم تقطیر میشوند. که به دو صورت امکان پذیر است:
1) روش مستقیم: که مخلوط آب و ماده آلی با همدیگر حرارت داده میشوند (تقطیر بوسیله آب).
2) روش غیر مستقیم: که بخار آب را در ظرف دیگری ایجاد کرده و از داخل ماده آلی عبور میدهند.
در تقطیر با بخار آب طبق قانون دالتون فشار بخارهای حاصله در درجه حرارت معین، برابر با مجموع فشارهای جزئی همان بخارها است:
PT = P1 + P2 + P3 + …
از این عبارت چنین بر می آید که همواره در هر درجه حرارتی فشار بخار کل مخلوط حتی از فشار بخار فرار ترین جزء در آن درجه حرارت بیشتر است، زیرا که فشار بخار اجزای دیگر مخلوط هم دخالت میکنند. بنابر این باید درجه جوش مخلوط ترکیبهای غیر قابل اختلاط کمتر از جزئی باشد که کمترین نقطه جوش را دارد.
آب (با نقطه جوش 100 درجه) و بروموبنزن (با نقطه جوش 156 درجه) در یکدیگر نامحلولند. این مخلوط در حدود 95 درجه سانتیگراد میجوشد. در این درجه، فشار بخار کل مخلوط برابر با فشار آتمسفر است. همانگونه که طبق نظریه دالتون پیش بینی میشد این درجه کمتر از نقطه جوش هر یک از این دو ماده به صورت خالص است.
مزیت استفاده از تقطیر با بخار آب در این است که در جه حرارت در این تقطیر نسبتا پایین است (کمتر از 100 درجه) و این روش برای خالص سازی موادی به کار میرود که نسبت به حرارت حساسند و در حرارتهای بالا تجزیه میشوند. همچنین این روش برای جدا کردن ترکیب، از مخلوط واکنشی که محتوی مقدار زیادی از مواد قیر مانند باشد مفید است.
برج تقطیر
برجهای تقطیر با سینی کلاهکدار ، تعداد سینیها در مسیر برج به نوع انتقال ماده و شدت تفکیک بستگی دارد. قطر برج و فاصله میان سینیها به مقدار مایع و گاز که در واحد زمان از یک سینی میگذرد، وابسته است. هر یک از سینیهای برج ، یک مرحله تفکیک است. زیرا روی این سینیها ، فاز گاز و مایع در کنار هم قرار میگیرند و کار انتقال ماده از فاز گازی به فاز مایع یا برعکس در هر یک از سینیها انجام میشود. برای اینکه بازدهی انتقال ماده در هر سینی به بیشترین حد برسد، باید زمان تماس میان دو فاز و سطح مشترک آنها به بیشترین حد ممکن برسد.
بخشهای مختلف برج تقطیر با سینی کلاهکدار
• بدنه و سینیها: جنس بدنه معمولا از فولاد ریخته است. جنس سینیها معمولا از چدن است. فاصله سینیها را معمولا با توجه به شرایط طراحی ، درجه خلوص و بازدهی کار جداسازی بر میگزینند. در بیشتر پالایشگاههای نفت ، برای برجهای تقطیر به قطر 4ft فاصله میان 50 - 18 سانتیمتر قرار میدهند. با بیشتر شدن قطر برج ، فاصله بیشتری نیز برای سینیها در نظر گرفته میشود.
• سرپوشها یا کلاهکها: جنس کلاهکها از چدن میباشد. نوع کلاهکها با توجه به نوع تقطیر انتخاب میشود و تعدادشان در هر سینی به بیشترین حد سرعت مجاز عبور گاز از سینی بستگی دارد.
• موانع یا سدها: برای کنترل بلندی سطح مایع روی سینی ، به هر سینی سدی به نام "وییر" (Wier) قرار میدهند تا از پایین رفتن سطح مایع از حد معنی جلوگیری کند. بلندی سطح مایع در روی سینی باید چنان باشد که گازهای بیرون آمده از شکافهای سرپوشها بتوانند از درون آن گذشته و زمان گذشتن هر حباب به بیشترین حد ممکن برسد. بر اثر افزایش زمان گذشتن حباب از مایع ، زمان تماس گاز و مایع زیاد شده ، بازدهی سینیها بالا میرود.
برجهای تقطیر با سینیهای مشبک
در برجهای با سینی مشبک ، اندازه مجراها یا شبکهها باید چنان برگزیده شوند که فشار گاز بتواند گاز را از فاز مایع با سرعتی مناسب عبور دهد. عامل مهمی که در بازدهی این سینیها موثر است، شیوه کارگذاری آنها در برج است. اگر این سینیها کاملا افقی قرار نداشته باشند، بلندی مایع در سطح سینی یکنواخت نبوده و گذر گاز از همه مجراها یکسان نخواهد بود.
خورندگی فلز سینیها هم در این نوع سینیها اهمیت بسیار دارد. زیرا بر اثر خورندگی ، قطر سوراخها زیاد میشود که در نتیجه مقدار زیادی بخار با سرعت کم از درون آن مجاری خورده شده گذر خواهد کرد. و میدانیم که اگر سرعت گذشتن گاز از حد معینی کمتر گردد، مایع از مجرا به سوی پایین حرکت کرده بازدهی کار تفکیک کاهش خواهد یافت.
برجهای تقطیر با سینیهای دریچهای
این نوع سینیها مانند سینیهای مشبک هستند. با این اختلاف که دریچهای متحرک روی هر مجرا قرار گرفته است. در صنعت نفت ، دو نوع از این سینیها بکار میروند:
1. انعطاف پذیر: همانطور که از نام آن برمیآید، دریچهها میتوانند بین دو حالت خیلی باز یا خیلی بسته حرکت کنند.
2. صفحات اضافی: در این نوع سینیها ، دو دریچه یکی سبک که در کف سینی قرار میگیرد و دیگری سنگین که بر روی سه پایهای قرار گرفته ، تعبیه شده است. هنگامی که بخار کم باشد، تنها سرپوش سبک به حرکت در میآید. اگر مقدار بخار از حد معینی بیشتر باشد، هر دو دریچه حرکت میکنند.
مقایسه انواع گوناگون سینیها
در صنعت نفت ، انواع گوناگون سینیها در برجهای تقطیر ، تفکیک و جذب بکار برده میشوند. ویژگیهایی که در گزینش نوع سینی برای کار معینی مورد توجه قرار میگیرد، عبارت است از: بازدهی تماس بخار و مایع ، ظرفیت سینی ، افت بخار در هنگام گذشتن از سینی ، زمان ماندن مایع بر روی سینی ، مشخصات مایع و ... . چون در صنعت بیشتر سینیهای کلاهکدار بکار برده میشوند، برای مقایسه مشخصات سینیهای دیگر ، آنها را نسبت به سینیهای کلاهکدار ارزیابی میکنند.
برجهای انباشته
در برجهای انباشته ، بجای سینیها از تکهها یا حلقههای انباشتی استفاده میشود. در برجهای انباشته حلقهها یا تکههای انباشتی باید به گونهای برگزیده و در برج ریخته شوند که هدفهای زیر عملی گردد.
1. ایجاد بیشترین سطح تماس میان مایع و بخار
2. ایجاد فضا مناسب برای گذشتن سیال از بستر انباشته
جنس مواد انباشتی
این مواد باید چنان باشند که با سیال درون برج ، میل ترکیبی نداشته باشند.
استحکام مواد انباشتی
جنس مواد انباشتی باید به اندازه کافی محکم باشد تا بر اثر استفاده شکسته نشده و تغییر شکل ندهد.
شیوه قرار دادن مواد انباشتی
مواد انباشتی به دو صورت منظم و نامنظم درون برج قرار میگیرند.
1. پر کردن منظم: از مزایای این نوع پر کردن، کمتر بودن افت فشار است که در نتیجه میشود حجم بیشتر مایع را از آن گذراند.
2. پر کردن نامنظم: از مزایای این نوع پر کردن ، میتوان به کم هزینه بودن آن اشاره کرد. ولی افت فشار بخار در گذر از برج زیاد خواهد بود.
مقایسه برجهای انباشته با برجهای سینیدار
در برجهای انباشته ، معمولا افت فشار نسبت به برجهای سینیدار کمتر است. ولی اگر در مایع ورودی برج ، ذرات معلق باشد، برجهای سینیدار بهتر عمل میکنند. زیرا در برجهای انباشته ، مواد معلق تهنشین شده و سبب گرفتگی و برهم خوردن جریان مایع میگردد. اگر برج بیش از حد متوسط باشد، برج سینیدار بهتر است. زیرا اگر در برجهای انباشته قطر برج زیاد باشد، تقسیم مایع در هنگام حرکت از بستر انباشته شده یکنواخت نخواهد بود.
در برجهای سینیدار میتوان مقداری از محلول را به شکل فرایندهای کناری از برج بیرون کشید، ولی در برجهای انباشته این کار، شدنی نیست. کارهای تعمیراتی در درون برجهای سینیدار ، آسانتر انجام میگیرد. تمیز کردن برجهای انباشته ، از آنجا که باید پیش از هرچیز آنها را خالی کرده و بعد آنها را تمیز نمایم، بسیار پرهزینه خواهد بود.
نکات مهم در انجام عمل تقطیر:
1-از دستگاه تقطیری که رابط های آن شل باشند ممکن است بخارهای قابل اشتعالی که باچراغ بونزن مجاور مشتعل شود (نشت کند). دستگاهی که گیره های آن سفت بسته شده باشند ممکن است ضمن کار آزمایشگاهی در اثر فشار به نقطه ی شکست خود برسد و علاوه بر خطرات فیزیکی که شیشه شکسته دارد باعث پخش مواد قابل اشتعال یا سوزان شود.
2-در بستن گیره باید دهانه گیره با قطعه شیشه ای که به آن بسته می شود به صورت موازی قرار گیرد. این حالت باعث می شود که گیره بدون کج کردن شیشه بسته شود وموجب شکستن شیشه یا شل کردن رابط دیگری نشود.قبل از اطمینان از وضعیت درست قطه ی شیشه ای و همترازی صحیح گیره آن را سفت نکنید.
3-در تقطیر ساده دستگاهی که به کار می رود در انتهای گیره خنک کننده به هوا راه داردو از این راه تعادل فشار برقرار می شود.در آزمایشگاه هیچگاه نباید یک دستگاه بسته را حرارت داد. چنان چهت تعادل فشاربرقرار نشود انبساط مواد در دستگاه فشار را زیاد ی کند و این عمل ممکن است باعث انفجار دستگاه شود.
4-در این آزمایشات باید توهجه داشت که موادی مثل بنزن و تولوئن باید خشک باشند.چنان چه از خشک بودن آنها اطمینان ندارید برای خشک کدن هر یک می توانید 50میلی لیتر از آن را در دستگاه تقطیرساده ای بریزید و تقطیر کنید تا مایع مقطر کدورتی را نشان ندهد.تقطیر را قطع کنید و باقی مانده تقطیر (نه مایع مقطر)را در آزمایش تقطیر جز به جز به کار برید.این روش برای خشک کردن حلال های مر طوبی مناسب است که با آب آزئو تروپ با جوشش مینی مم می دهند.
5-هنگامیکه مایع قابل اشتعالی را در حالت تقطیر یا رفلاکس حرارت می دهید اطمینان حاصل کنید که تمام رابط ها نحکم و عاری از فشار باشند.در موقعی که مایع بسیار فراری را گرم می کنید بهتر است که یک لوله لاستیکی را به دهانه باز دستگاهی متصل می کنید و لوله را از لبه بالای میز کار بگذرانید.و آن را از چرا گاز خود دور کنید.
6-هرگز نگذارید که یک محصول تقطیر قابل اشتعال به خصوص اگه نزدیک به شعله نفر پهلویی شما باشد آزادانه از خنک کننده به ظرف گیرنده ای که چند اینچ پایین تر از آن است بچکد.برای هدایت محصول تقطیر به ظرف گیرنده از یک مبدل استفاده کنید.
ضمیمه:
حلالهای قابل اشتعال که معمولا با آنها زیاد سرو کار داریم به ترتیب در زیر آورده شده است:
1-اتیل اتر
2-استرها(اتیل استات)
3-الکلها(متانول-اتانول-2-پروپانول)
4-کتون ها(استون و بوتانون)
5-کربن دی سولفید
6-هیدروکربن ها(پنتان -هگزان-بنزن-تولوئن و...)
منبع:
1)کتاب شیمی آلی تجربی نوین-جلد اول و جلد دوم-نام نویسندگان:رابرتس-گیلبرت-ردوالد-وینگرو-نام مترجم:هوشنگ پیر الهی
2)کتاب شیمی عملی و آلی-مولفین:آقایان جلیلیان-وارسته مرادی-احمدی گلسفیدی
3)technique of chemistry/A.Weissberger
سایت های مرتبط:
1)http://www.daneshnamehroshd.com/
2)http://chemlab.mihanblog.com/
3)http://www.distillationgroup.com/distill.htm
4)http://orgchem.colorado.edu/hndbksupport/dist/dist.html
واکنش های یون کوییوریک ومرکوریک وبیسموت وکادمیم
واکنش های Cu2+(یون کوِییوریک)
برای بررسی این کاتیون ها از محلول سولفات مسll استفاده میشود.
1. اثر :H2S
رسوب سیاه رنگ،سولفورمس در محیط HCLرقیق حاصل می شود که در اسید نیتریکگرم محلول بوده امادر اسید گرم،غلیظ
وغیر محلول است و در هیدروکسید های قلیایی نیز نا محلول است.
تذکر:برای تهیه گاز H2S در آزمایشگاه،محلول یتو استامیدو HCLرقیق به لوله آزمایش محتوی کاتیون مربوطه اضافه کنید
وبه ملایمت حرارت دهید.
CuSO4+H2S→CuS+H2SO4
2. اثر محلول سود:
رسوب هیدروکسید مس آبی رنگ،تولید می شود.که در مقدار اضافی معرف نا محلولاست ولی در اثر حرارت دادن به اسید مس
سیاه رنگ تبدیل میشود.
Cu (SO) 4+2NaOH→Cu (OH) 2+2Na+SO
حرارت
Cu (OH) 2→CaO+H2O
3. اثرمحلول آمونیاک:
دراضافی معرف،کمپلکس آمونیاکی مسll بارنگ آبی پررنگ می دهد.
CuSO4+4NH3→ [Cu (NH3)4] SO4
4. اثرمحلول فروسیانور پتاسیم:
رسوب قرمز قهوه ای رنگ،فروسیانور مس میدهد.
K2Fe [Fe (CN6)] +2CuSO4→Cu2 [Fe (CN6)] +FeK2 (SO4)
5. اثر محلول یدور پتاسیم:
ابتدا یدور مسllرسوب میکند که بلافاصله در حضور یدور اضافی به CuI سفید رنگ وI2آزاد تبدیل میگردد کهرنگ محلول را
قهوه ای می کند.
CuSO4+KI→CuI+KSO4
واکنش های یون مرکوریک:
برای بررسی این کاتیون ها از محلول کلرید یا نیترات جیوۀll استفاده می شود.
1. اثرH2S:
ابتدارسوب سفید،بعدزرد،سپس قهوه ای ودر آخر سیاه رنگ،HgS می دهد.ترکیباترنگی که به علت ناکافی بودنH2S تولید میشوند،
ترکیباتی با نسبت های مختلفS2-،Cl-،Hg2+می باشند.که در نتیجه واکنش کافی H2S تماماًبه HgS تبدیل میشوند.
HgCl2+H2S→HgS+2HCl
HgS+H2S→HgS2+H2
HgS2+H2S→HgS+2HS
2. اثر محلول SnCl2:
ابتدا رسوب سفید رنگ،Hg2Cl2 میدهد که در اثر اضافی معرف به جیوۀ سیاه رنگ تبدیل میشود.
2HgCl2+SnCl2→Hg2Cl2+SnCl4
Hg2Cl2+SnCl2→Hg+SnCl4
3. اثر محلول سود:
رسوب زرد رنگ،اکسید جیوۀll می دهد.
HgCl2+2NaOH→HgO+NaCl+H2O
4. اثر محلول آمونیاک:
برخلاف Hg22+،جیوۀ فلزی آزاد نمی کند فقط رسوب سفید رنگ،HgNH2Cl تولید میشود.
HgCl2+NH3→HgNH2Cl+NHCl
5. اثر محلول یدور پتاسیم:
ابتدا یدور جیوۀll قرمز آجری می دهد که در اضافی معرف با تشکیل کمپلکس حل میگردد.این کمپلکس معرف نسلر نام دارد که
برای شناسایی املاح آمونیوم به کار میرود.
HgCL2+2KI→HgI22KCl
HgI2+2KI→K2 [HgI4] تترا مرکوراتمعرف نسلر
واکنش های یون بیسموت
واکنش های یون بیسموتاز محلول نبترات بیسموت استفاده می شود.
1. اثرH2S:
رسوب قهوه ای رنگ،سولفور بیسموت حاصل میشود که در اسیدهای رقیق و سردو همچنین در هیدروکسیدها غیر محلول است.
ولی در اسید نیتریک گرم محلول است.
Bi2S3+8HNO3→2Bi (NO3)2+2NO+3S↓+4H2O
2. اثر محلول سود:
رسوب سفید رنگ،هیدروکسید بیسموت تولید میگردد.Bi(OH)3
BiNO3+3NaOH→Bi (OH) 3+3Na+3NO
3. اثر محلول یدید پتاسیم:
رسوب قهوه ای تیره،یدید بیسموت می دهد.به یک قسمت ازرسوب مقدار بیشتر معرف ریخته ورسوب باتشکیل کمپلکس[BiI4]نارنجی
رنگ حل میشود.
BiI3+I-→ [BiI4]-
به قسمت دیگر رسوب آب مقطر اضافه کنید.دررقّت زیاد رسوب پرتقالی رنگ،یدور بیسموتیل می دهد.
BiI3+H2O→BiOI↓+2HI
4. اثر کلرید قلع ll:(کلرید استانو)
در محیط قلیایی رسوب سیاه رنگ،بیسموت فلزی می دهد.
2Bi (NO3)3+3SnCl2+18NaOH→2Bi↓+3Na2 [Sn (OH) 6] +6NaNO3+6NaCl
واکنش های یون کادمیم(Cd2+)
از محلول سولفات کادمیم استفاده می شود.
1. اثرH2S:
رسوب زرد رنگ، سولفور کادمیم می دهد که در اسید سولفوریک گرم واسید نیتریک گرم محلول است.
Cd2++H2S→CdS↓+2H+
CdS+H2SO4→CdSO4+H2S
2. اثر محلول سود:
رسوب سفید رنگ،هیدروکسیدکادمیم می دهد که در اضافی معرف غیر محلول است.
2OH-+Cd2+→Cd (OH) 2↓
3. اثر محلول آمونیاک:
ابتدا رسوب سفید رنگ،هیدروکسید کادمیم می دهد که در اضافی معرف غیر محلول است.
Cd(OH)2+4NH3→[Cd(NH3)4](OH)2
گاز سنتزی
اصطلاح گاز سنتز به مخلوطهای گازی اطلاق میشود که محتوی منوکسیدکربن و هیدروژن به نسبتهای مختلف باشند. هیدروژن و منوکسیدکربن دو مادة مهم در صنایع شیمیایی محسوب شده و دارای مصارف و کاربردهای فراوانی میباشند. منوکسیدکربن در تولید رنگها، پلاستیکها، فومها، حشرهکشها، علفکشها، اسیدها و ... به کار میرود. از جمله مصارف هیدروژن نیز میتوان به تولید آمونیاک، هیدروژناسیون و هیدروکراکینگ اشاره نمود.
گاز سنتز مادة اولیه بسیار با ارزشی جهت تولید مواد متنوع شیمیایی میباشد. با استفاده از این گاز و فرایندهای مختلف، میتوان مواد متنوع شیمیایی را تولید نمود که بسته به روش تولید آن نسبتهای مختلف هیدروژن به منوکسیدکربن به دست میآید. همچنین در موارد مصرف در صنعت، بسته به فرایندی که گاز در آن مورد استفاده قرار میگیرد، نسبتهای مختلف لازم است.
موارد مصرف گاز سنتز عمده موارد مصرف گاز سنتز به شرح ذیل است:
از آنجاییکه متانول به مقدار زیاد در سنتز استیک اسید مصرف میشود، اهمیت فراوانی در صنعت دارد.
در این نوع واکنشها از اولفینها با استفاده از گاز سنتز، آلدئید تولید میشود. این واکنش اکسو سنتز نیز نامیده میشود.
در این فرایند گاز سنتز به مولکولهای بنزینی در گستره تبدیل میشود. در اصل این واکنش اولیگومریزاسیون منوکیسدکربن به وسیلة هیدروژن جهت تشکیل محصولات آلی میباشد.
جهت احیای سنگ آهن به دست آمده از معادن، از گاز سنتز استفاده میشود در این فرایند آهن یا پودر آن به وسیله احیای مستقیم کانیهای آهن به دست میآیند.
از جمله دیگر مصارف گاز سنتز، میتوان به تهیه الکلهای سنگین، دیمتیل اتر، استرها، کتونها، هیدروکربورها و غیره اشاره کرد.
روشهای تهیة گاز سنتز
این روش، اولین روش تولید گاز سنتز است که در آن گاز سنتز توسط گازی شکل کردن کک از ذغال سنگ در دماهای پایین به وسیلة هوا و بخار آب به دست میآید:
این فرایند غیر کاتالیستی بوده و نسبت تولیدی توسط آن کم، و در حدود 1 است. با توجه به وجود مواد متنوع در ذغال سنگ، گاز سنتز تولیدی از این روش نیازمند واکنشها و خالصسازیهایی جهت تولید گاز سنتز با خلوص بالا میباشد.
این فرایند، غیرکاتالیستی بوده و در اصل احتراق جزئی هیدروکربن در حضور اکسیژن و بخار آب میباشد. موقعی که متان به عنوان خوارک مورد استفاده قرار گیرد، مزیت عمدة این روش که یک فرایند تولید گرما میباشد این است که طیف گستردهای از هیدروکربنها را به عنوان خوراک میتواند مورد استفاده قرار دهد. ترکیب گاز سنتز تولیدی بستگی به نسبت کربن به هیدروژن خوراک و مقدار بخار اضافه شده دارد.
این فرایند واکنش کاتالیستی هیدروکربن و عامل تغییر شکل دهنده (Reforming agent ) در دمای بالا میباشد. عامل تغییر شکل دهنده میتواند بخار آب، دیاکسید کربن، اکسیژن و یا مخلوط آنها باشد. ترکیب درصد گاز سنتز تولیدی بستگی به نوع هیدروکربن به کار رفته، عامل تغییر شکل دهنده و مقدار آن، شرایط عملیاتی و نوع کاتالیست دارد ۱- تهیة متانول ۲- تهیة اتیلن گلیکول ۳- واکنشهای هیدروفرمیلدار کردن ۴- سنتز فیشر- تروپش ۵- احیای سنگ آهن ۶- سایر مصارف 1- گازیشکلکردن زغال سنگ ۲- اکسیداسیون جزئی هیدروکربنها ۳- رفرمینگ هیدروکربنها
چگونگی کشف هیدروژن
هیدروژن یکی از جالب ترین عناصر جدول تناوبی است عدد اتمی اش یک و سبک ترین گاز موجود در طبیعت است عنصری است که برای حل بسیاری از مسائل شیمی نظری کشفش ضروری بوده عنصری است که با از دست دادن تنها الکترونش تبدیل به پروتونی عریان و به دون پوشش می شود و بنابراین شیمی هیدروژن شیمی ویژه ای است و در واقع شیمی یکی از ذرات اساسی است.
مندلیف هیدروژن را عادی ترین عنصر درمیان عناصر عادی می نامد (وی عناصر موجود در تناوبهای کوتاه جدول تناوبی را عادی تلقی می کرد) زیرا این عنصر آغازگر سری عناصر شیمیایی طبیعی بود می توان با واکنش ساده مانند ریختن اسید کلرئیدریک بر روی براده روی مقداری هیدروژن تهیه کرد.
حتی در دورانهای کهن که هنوز شیمی به عنوان علم تلقی نمی شد و کیمیا گران در جستجوی کیمیا بودند اسید کلرئیدریک، اسید سولفوریک،اسید نیتریک،آهن وروی ، شناخته شده بودند به عبارت دیگر بشر کلیه موادی را که با اثر دادنشان بر هم می توانست هیدروژن تهیه کند در اختیار داشت . تنها برای شناختن آن می بایستی واقعه ای رخ دهد . در نوشته های قرنهای دهم تا دواردهم شمسی / شانزدهم تا هیجدم میلادی گزارشهایی موجود است که نشان می دهد در برخی موارد مانند ریختن اسیدسولفوریک بر روی براده آهن گازی متساعد می شده است که در آن زمان تصور می شد که نوعی هوای قابل اشتعال است . یکی از کسانی که به این نوع هوای اسرار آمیز برخورده است لومونوزوف دانشمند مشهور روسی بوده است . در سال 1124/ 1745 وی رساله ای تحت عنوان « درباره درخشندگی فلزی» نوشت که از جمله مطالبش یکی این بود که :« با حل کردن برخی فلزات پست ، به ویژه آهن ، در الکل اسیدی شده ،بخارات قابل اشتعال از دهانه باز دستگاه آزمایش خارج می شود .... (بنابر اصطلاحات متداول آن زمان ، اسید را الکل اسیدی شده می نامند ) بنابراین ، آنچه که لومونوزوف دیده بود چیزی جز هیدروژن نبوده است . ولی اگر جمله اش را تا آخر بخوانیم ، می بینم که نوشته است ... این بخارات فلوژیستون است . نظر به اینکه انحلال فلز در اسید موجب تولید materia ignea یا بخاری قابل اشتعال می شد ، خیلی مناسب بود که آن را به اینگونه تفسیر کنند که انحلال فلز ، سبب آزاد شدن فلوژیستون می شود . با این عبارت پردازی ، واکنش مزبور منطبق بر « نظریه آتش زایی » می شود . اینک به جا است که با کاوندیش دانشمند ارزنده انگلیسی آشنا شویم وی با تعصب عجیبی به علم گرایش داشت و آزمایشگر برجسته ای بود . وی هرگز در انتشار نتایج تجربه هایش عجله نداشت و گاه سالها می گذشت تا مطلبی منتشر کند بنابراین مشکل است بتوان به طور دقیق معلوم کرد که وی آزاد شدن « هوای قابل اشتعال» را در چه تاریخ مشاهده کرده است .
نکته ای که در این مورد معلوم است ، انتشار مطلبی در سال 1145 شمسی / 1766 میلادی تحت عنوان «آزمایشهایی با هوایی مصنوعی» بود که یکی از موضوعات اساسی را در پژوهش های شیمی هوایی تشکیل داد . در عین حال به نظر می رسد که آن تجربیات بر اثر پافشاری بلاک انجام شده باشد . « هوای ثابت » توجه کاوندیش را جلب کرده بود و در نتیجه وی تصمیم گرفته بود ببیند آیا نوع دیگری از هوای مصنوعی وجود دارد یا خیر . در این بررسیها او اشاره به نوع دیگری از هوا می کرد که در ترکیبات وجود دارد و به طور مصنوعی قابل جدا کردن از آنها است . ولی می دانست که هوای قابل اشتعال را درموارد متعدد دیده اند و خودش هم به همان روش یعنی اثر دادن اسید سولفوریک و اسید کلرئیدریک بر آهن ، روی و قلع ، به تهیه آن مبادرت ورزید . با انجام این آزمایشها ،وی نخستین فردی بود که ثابت کرد که در همه موارد یاد شده نوع مشابهی هوا یعنی «هوای قابل اشتعال » را مورد توجه قرار می گیرد . کاوندیش به عنوان پیرو «نظریه آتش زایی » تنها به یک نوع تفسیر در باره طبیعت ماده اعتقاد داشت . بنابراین او هم مانند لومونوزوف، این ماده را فلژیستون نامید . وقتی وی درباره خواص «هوای قابل اشتعال » بررسی می کرد ، مطمئن بود که مشغول بررسی خواص «فلژیستون » است . او ضمن کارهایش به این نتیجه رسیده بود که فلزات مختلف ، حاوی نسبت های مختلفی از «هوای قابل اشتعال » هستند .بنابراین به «هوای ثابت »مورد اداعای بلاک ، «هوای قابل اشتعال » کاوندیش هم اضافه شد . به طور خلاصه آن دو دانشمند چیز تازه ای کشف نکرده اند و فقط توانستند مشاهدات گذشتگان را جمع بندی کنند . اما همین جمع بندیها سبب پیشرفت چشمگیری در تاریخ دانش بشری شد .«هوای ثابت » و «هوای قابل اشتعال» با یکدیگر و با هوای معمولی تفاوت داشتند.«هوای قابل اشتعال » به طور اعجاب آور سبک وزن بود .کاوندیش متوجه شد که فلوژیستونی که به دست آورده است ، دارای جرم است. وی نخستین فردی بود که کمیت چگالی را برای اندازه گیریهای مربوط به گازها معرفی کرد . وقتی چگالی هوا را برابر واحد فرض کرد ، برای چگالی «هوای قابل اشتعال» مقدار 9./. و برای چگالی «هوای ثابت » مقدار 75/1 بدست آورد . اما در اینجا میان کاوندیش تجربه گر و کاوندیش طرفدار «نظریه آتش زایی» اختلاف بروز کرد زیرا با توجه به این که «هوای قابل اشتعال » دارای جرم است ،به همین وجه نمی توان آن را فلوژیستون خالص تلقی کرد .به عبارت دیگر فلزاتی که هوای قابل اشتعال را از دست می دهند ، اجبارا باید دستخوش کم شدن جرم هم بشوند کاوندیش برای رفع این تناقض ، فرضیه ای بی محتوا به این شرح ابراز داشت :«هوای قابل اشتعال » مجموعه ای از فلوژیستون و آب است . حاصل آن فرضیه این بود که بلاخره در ترکیب «هوای قابل اشتعال » هیدروژن ظاهر شد .
نتیجه آشکار این است که گر چه کاوندیش «هوای قابل اشتعال »را وزن کرد ، خواصش را شرح داد و آن را نوع ویژه ای از «هوای مصنوعی » دانست ،ولی او هم مانند پیشینیانش به ماهیت این ماده پی نبرد . به عبارت دیگر کاوندیش به دون آگاهی از واقیعت ماده ای را که به دست آورده بود به عنوان فلوژیستون مورد مطالعه قرار داد ، نه به عنوان عنصر شیمیایی جدید و علت این اشتباه ، پایبند بودنش به «نظریه آتش زایی » بود . وقتی وی متوجه شد که خواص «هوای قابل اشتعال » مغایر با آن نظریه است «فرضیه ای که به اندازه نظریه گفته شده گمراه کننده بود ، ارائه داد.
بنابراین اگر بگوییم «هیدروژن را کاوندیش دانشمند انگلیسی در سال 1145 /1766 کشف کرده است» ، حرف نادرستی زده ایم . درمقایسه با دیگران ، کاوندیش روشهای تهیه و نیز خواص « هوای قابل اشتعال » را با جزئیات بیشتری شرح داده است . در هر حال ولی در عین حال نمی دانست چکار می کند و طبیعت عنصری هوای قابل اشتعال بر او روشن نشده بود . ولی نمی توان گناه را به گردن این دانشمند گذاشت ، بلکه باید گفت که شیمی هنوز به آن درجه از کمال که چنین پیشبینیهایی داشته باشد ،نرسیده بود . سالها گذشت تا سرانجام هیدروژن واقعا هیدروژن شد و جای شایسته اش را در شیمی اشغال کرد نام لاتین آن hydrogcnium از دو کلمه یونانی hydro وgcnnac به معنی « آبزا » گرفته شده است . این نام را لااووازیه درسال 1158 / 1779 پس از معلوم شدن ترکیب آب ،پیشنهاد کرد . حرفH به عنوان علامت شیمیاییش توسط برزلیوس پیشنهاد شده است . هیدروژن از جهت اینکه ایزوتوپهایش هم از نظر خواص فیزیکی با هم متفاوتند و هم از نظر خواص شیمیایی ، عنصری منحصر به فرد است . زمانی این تفاوت ها برخی دانشمندان را واداشت که ایزوتوپهای هیدوژن را به عنوان عناصر جداگانه ای تلقی کنند و برایشان جای ویژه ای در جدول تناوبی پیدا کنند .بنابراین تاریخچه کشف ایزوتوپهای هیدروژن همانند تاریخچه خود هیدروژن جالب توجه است .
جستجوی ایزتوپهای هیدروژن در دهه اول قرن حاضر شمسی /دهه سوم قرن حاضر میلادی آغاز شد ولی همه کوششها ناکام ماند و نتیجه گیری کردند که هیدروژن ایزوتوپ ندارد . در سال 1310/ 1931 پیشنهاد شد که هیدروژن باید دارای ایزوتوپی به عدد جرمی 2 باشد . نظر به اینکه چنین ایزوتوپی جرمش دو برابر هیدروژن است ، دانشمندان برای جدا کردن هیدروژن سنگین به روشهای فیزیکی متوسل شدند. در سال 1311 /1932 سه نفر دانشمند آمریکایی به نامهای یوری ، بریکود و مورفی هیدروژن مایع را تبخیر کردند و با مطالعه باقی مانده تبخیر به روش طیف سنجی ،ایزوتوپ سنگین را در آن یافتند .وجود هیدروژن سنگین در هوای جو در سال 1320/1941محقق شد.نام دوتریوم deuterium از کلمه یونای deuteron به معنی «دومین یا یکی دیگر » گرفته شده است . ایزتوپ دیگر با عدد جرمی سه به نام تری تیوم( مشتق از کلمه یونانی tritos به معنی سومین ) رادیواکتیو است و در سال 1313 /1934 توسط سه دانشمند انگلیسی به نامهای اولیفانت ،هارتک و رادرفوردشناخته شده است . به ایزوتوپ اصلی هیدروژن نام پروسیوم نیز داده اند .این تنها موردی از که ایزوتوپهای یک عنصر نامها و علامتهای شیمیایی متفاوت دارند (hوt و d ).99/99 درصد همه هیدروژنها از پروسیوم است و بقیه اش دوتریوم می باشد . به صورت اثر بسیار ناچیزی همواره دو ایزوتوپ دیگر است .
نیکل و تاثیرات آن بر انسان
فلز نیکل
نیکل فلزی سخت ، چکش خوار، براق با ساختار بلورین مکعبی به رنگ سفید- نقره ای است . این عنصر در سال 1751 توسط Axel Cronstedt دانشمند سوئدی کشف گردید . از نظر خواص مغناطیسی وفعالیت شیمیایی شبیه به آهن وکبالت است . کانیهای اصلی نیکل پنتلاندیت ، پیروتیت (سولفید های نیکل- آهن) و گارنییریت (سیلیکات نیکل- منیزیم ) هستند.
نیکل یکی از اجزا اصلی بیشتر شهابسنگها به شمار می آید. شهابسنگهای آهن و سیدریت شامل آلیاژهای آهن حدود 5 تا 20 درصد نیکل می باشد. نیکل تجاری به فرمهای پنتلاندیت و پیروتیت می باشد که این معادن در ایالت انتاریو یافت می شود که این ناحیه حدود 30 درصد از نیکل دنیا را تامین می کند. دیگر معادن این عنصر در کالندونیا، استرالیا، کوبا، اندونزی و در مناطق دیگر یافت می شود. این عنصر رسانای جریان بر ق است و سطح آن براق و صیقلی می باشد. اینعنصر از گروه عناصر آهن و کبالت می باشد و آلیاژهای آن قیمتهای بالایی دارند. این عنصر کاربردهای فراوانی در طبیعت دارد و برای ساخت فولاد ضدزنگ و دیگر آلیاژهای ضد زنگ و خوردگی مثل اینوار و مانل که الیاژى از نیکل و کبالت که در برابر خوردگى مقاوم است و و اینکونل و Hastelloys کاربرد دارد. برای ساخت لوله های نیکلی و مسی و همینطور برای نمک زدایی گیاهان و تبدیل آب شور به آب مایع استفاده می شود. نیکل استفاده های فراوانی برای ساخت سکه ها و فولاد نیکلی برای زره ها و کلید ها کار برد دارد و همینطور از نیکل می توان آلیاژهای نیکروم و پرمالوی و آلیاژی از مس را تهیه کرد. از نیکل برای ساخت شیشه های به رنگ سبز استفاده می شود. صفحات نیکلی می تواند نقش محافظت کننده برای دیگر فلزات را داشته باشد. نیکل همچنین کاتالیزوری برای هیدروژن دار کردن روغنهای گیاهی است. همچنین صنعت سرامیک و ساخت آلیاژی از آهن و نیکل که خاصیت مغناطیسی دارد و باتری های قوی ادیسون کاربرد دارد. از ترکیبات مهم نیکل می توان سولفات و آکسید را نام برد. نیکل طبیعی مخلوطی از 5 ایزوتوپ پایدار است . همچنین 9 ایزوتوپ ناپایدار دیگر نیز شناخته شده است. نیکل هم به صورت فلز و هم به صورت ترکیب محلول می تواند وجود داشته باشد. بخار سولفید نیکل سرطان زا می باشدکه در موقع استفاده از آن باید دقت لازم را به عمل آورد.
نیکل و تاثیرات آن بر انسان نیکل یکی از فراوانترین عناصر است. نیکل در طبیعت معمولا در ترکیب با اکسیژن (اکسیدها) یا گوگرد (سولفیدها) وجود دارد. یکل برای حفظ سلامت حیوانات ضروری است. با اینکه هیچ اثری در نتیجه کمبود نیکل در انسان دیده نشده است ولی احتمالا مقدار کمی از آن برای سلامتی انسان ضروری است ● نیکل:
اثرات نیکل بر سلامت انسان
مقدارنیکل در طبیعت بسیار کم است. انسان در زمینه های مختلف از نیکل استفاده میکند. یکی از عمده ترین کاربردهای نیکل، در صنعت فولاد است. از نیکل به عنوان یکی از اجزا سازنده فولاد و سایر محصولات فلزی استفاده میشود. حتی از نیکل در جواهرات هم استفاده میشود. مواد غذایی به طور طبیعی دارای مقداری نیکل هستند. شکلات و چربی ها دارای مقدار بسیار زیادی نیکل هستند. در صورتیکه افراد از سبزیجات حاصل از مناطق آلوده به نیکل تغذیه کنند، مقدار زیادی نیکل وارد بدنشان میشود. نیکل در بافت گیاهان تجمع می یابد و در نتیجه مقدار نیکل در سبزیجات افزایش پیدا میکند. در ششهای افراد سیگاری مقدار زیادی نیکل وجود دارد. همچنین نیکل در شوینده ها نیز مورد استفاده قرار میگیرد.راههای ورود نیکل به بدن انسان از طریق هوا، آشامیدن آب، خوردن غذا و کشیدن سیگار است. ممکن است بر اثر تماس پوست با خاک یا آب آلوده به نیکل، مقداری نیکل وارد بدن انسان شود. مقدار اندک نیکل برای انسان ضروری است اما اگر مقدار آن افزایش یابد، برای سلامت انسان خطرناک است. نتایج مصرف بالای نیکل به شرح زیر است:شانس مبتلا شدن به سرطان ریه، سرطان بینی، سرطان حنجره و سرطان پروستات را افزایش میدهد. پس از اینکه فرد در معرض گاز نیکل قرار گرفت، دچار کسالت و سرگیجه میشود. آب آوردن ریه ها
مشکلات تنفسی
کاهش توانایی تولید مثل
آسم و برونشیت مزمن
حساسیتهایی از قبیل خارش پوست (به خصوص در هنگام استفاده از جواهرات)نارسایی قلبی
بخارات نیکل به دستگاه تنفس و ریه ها آسیب میرساند. نیکل و ترکیبات آن باعث آماس پوست میشوند که تحت نام " خارش نیکل" نامیده میشود و معمولاً در افراد با حساسیت پوستی بالا مشاهده میشود. اولین علامت، خارش است که معمولاً هفت روز قبل از از بین رفتن پوست رخ میدهد. اولین علائم تخریب پوستی التهاب پوست یا پوسته پوسته شدن پوست است. سپس در پوست زخمهایی نمودار میشود. از لحاظ تقسیم بندی برنامه سمشناسی ملی آمریکا (NTP)، نیکل و ترکیبات آن جزعوامل سرطانزا محسوب میشوند و از نظر طبقه بندی آژانس بین المللی تحقیقات سرطان (IARC) ترکیبات نیکل در گروه یک قرار میگیرند. گروه یک شامل عناصری میباشد که شواهد کافی در مورد سرطانزایی آنها وجود دارد. در این تقسیم بندی عنصر نیکل در گروه 2B قرار دارد. گروه 2B عناصری هستند که ممکن است در انسان سرطان ایجاد کنند.
تاثیرات زیست محیطی نیکل کارخانه ها و سوزاندن زباله ها دو عامل اصلی در تولید نیکل و ورود آن به هوا میباشند. مقدار نیکلی که در هوا وجود دارد به مراتب از نیکل موجود در زمین بیشتر است. مدت زمان از بین رفتن نیکل موجود در هوا زیاد است. زمانیکه هرزآبها جریان پیدا میکنند، مقداری نیکل را وارد آبهای سطحی میکنند. بخش اعظم ترکیبات نیکل در طبیعت جذب ذرات خاک و رسوبات شده و در نهایت به صورت غیر متحرک درمی آیند. در زمینهای اسیدی نیکل بسیار متحرک میشود و معمولاً در آبهایزیرزمینی شسته میشود. شواهد چندانی درباره تاثیر نیکل بر سایر موجودات زنده به غیر از انسان وجود ندارد. در حال حاضر دانشمندان می دانند که غلظت بالای نیکل در خاکهای ماسه ای به گیاهان صدمه میزند و همچنین غلظت بالای نیکل در آبهای سطحی سبب کاهش تعداد و رشد جلبکها میشود. رشد موجودات ذره بینی نیز در حضور نیکل کاهش پیدا میکند، اما معمولاً با گذشت زمان در برابر نیکل مقاوم میشوند. مقدار اندک نیکل باید در غذای جانوران وجود داشته باشد. اما زمانیکه مقدار نیکل از حد مجاز خود فراتر رود، میتواند برای جانوران مضر و خطرناک باشد. جانورانی که در نزدیکی پالایشگاه زندگی میکنند، بر اثر دریافت مقدار زیاد نیکل به انواع مختلف سرطان مبتلا میشوند. از آنجاییکه نیکل در بافتهای گیاهی و جانوری نمیتواند تجمع پیدا کند، اثری در زنجیره غذایی ندارد.
تجهیزات آزمایشگاهی مورد استفاده در تجزیه
اسپکترومتر جرمی ، میکروسکوپ ، کرماتوگرافی مایع و گازی ، اشعه x ، جذب اتمی ، مادون قرمز ، کروماتوگرافی مایع با عملکرد بالا و اسپکترومتر نشری
خواص فیزیکی و شیمیایی عنصر نیکل :
عدد اتمی: 28
جرم اتمی:58.6934
نقطه ذوب: C°1435
نقطه جوش : C°2732
شعاع اتمی : Å 1.62
ظرفیت:2و3
رنگ: سفید – نقره ای
حالت استاندارد: جامد
نام گروه: 10
انرژی یونیزاسیون : Kj/mol 7.635
شکل الکترونی: 2 1s22s2p63s23 p63d 84s
شعاع یونی : Å 0.69
الکترونگاتیوی:1.91
حالت اکسیداسیون:2و3
دانسیته: 8.9
گرمای فروپاشی : Kj/mol 17.47
گرمای تبخیر : Kj/mol 370.4
مقاومت الکتریکی : Ohm m: 0.0000000699
گرمای ویژه: J/g Ko 0.44
دوره تناوبی:4
درجه اشتعال : در حالت جامد اشتعال پذیر
نیکل یکی از فراوانترین عناصر است. نیکل در طبیعت معمولا در ترکیب با اکسیژن (اکسیدها) یا گوگرد (سولفیدها) وجود دارد. این فلز در همه خاکها وجود دارد و از آتشفشانها نیز نشر می شود. نیکل خالص، فلزی سخت و به رنگ سفید-نقره ای است که با دیگر فلزات برای تشکیل آلیاژها ترکیب می شود. تعدادی از فلزات که با نیکل آلیاژ می شوند عبارتند از آهن، مس، کروم و روی .این آلیازها در ساخت سکه های فلزی، جواهرات و اجناس فلزی مورد استفاده قرار می گیرند.ترکبات نیکل همچنین در آبکاری نیکل، سرامیکهای رنگی، بعضی از باطریها و همچنین به عنوان کاتالیزور برای افزایش سرعت واکنشها بکار می روند. نیکل و ترکیباتش بو مزه خاصی ندارند.نیکل برای حفظ سلامت حیوانات ضروری است. با اینکه هیچ اثری در نتیجه کمبود نیکل در انسان دیده نشده است ولی احتمالا مقدار کمی از آن برای سلامتی انسان ضروری است. در محیط، نیکل بیشتر در خاک و رسوبات وجود دارد زیرا نیکل با ذراتی که حاوی آهن یا منگنز هستند و در خاکها و رسوبات موجود هستند، اتصال برقرار می کند.آژانس حفاظت از محیط زیست (EPA)، حداکثر مقدار مجاز نیکل در آب آشامیدنی کودکان را ۰۴/۰ میلی گرم در لیتر تعیین کرده است. میزان مجاز نیکل در هوای محل کارهای مرتبط، یک میلی گرم در مترمکعب برآورد شده است. در حال حاضر مقدا نیکل موجود در محیطهای کار، بسیار کمتر از گذشته است و به همین دلیل علائم آلودگی با نیکل در کارگران کمتر دیده می شود.منابع اصلی آلودگی با نیکل استعمال تنباکو، اگزوز خودرها، کودهای شیمیایی، سوپر فسفاتها، فرآورده های غذایی، روغنهای هیدروژنه، فاضلابهای صنعتی، صنایع فولاد زنگ نزن، آزمایش تجهیزات هسته ای، بکینگ پودر و ... می باشند. تنفس هوا یا دود تنباکوی محتوی نیکل و یا خوردن مواد غذایی و آب حاوی نیکل و تماس با سکه ها و فلزات حاوی نیکل، منابع اصلی آلودگی انسان با نیکل هستند.● تاثیرات نیکل بر انسان:
متداولترین اثر نیکل بر انسان یک واکنش آلرژیک است. انسان می تواند در صورت آلودگی با منابع ذکر شده در بالا دچار حساسیت شود. اشخاصی که به نیکل حساس هستند، در صورت تماس زیاد با آن دچار یک واکنش می شوند و معمولترین واکنش، تحریک آن قسمت از پوست است که با نیکل تماس پیدا کرده است. در برخی موارد ممکن است فرد حساس، در صورت آلودگی با نیکل دچار تنگی نفس می شوند. در کارگرانی که مقادیر بالایی از نیکل را تنفس کرده بودند مشکلات ریوی، شامل برونشیت مزمن و کاهش توان ریه ها مشاهده گردید.مسمومیت حاد با استنشاق نیکل کربونیل اتفاق می افتد. این اثرات حاد در طی دو مرحله ظاهر می شوند، مرحله اول اثرات فوری و مرحله دوم با اثرات با تاخیر. سردرد، سرگیجه، تنفس بریده بریده، تهوع و استفراغ علائم اولیهء آلودگی شدید است. اثرات تاخیری (۱۰ تا ۳۶ ساعت بعد) ظاهر می شوند و شاملِ درد سینه، سرفه، تنفس بریده بریده، بی رنگی و مایل به آبی شدن پوست و در موارد بسیار حاد, هذیان گویی، تشنج و مرگ می باشد. بهبودی این مسمومیت، طولانی خواهد بود. کارگرانی که بطور تصادفی آب آشامیدنی را که حاوی ۱۰۰٫۰۰۰ برابر حد مجاز نیکل را مصرف کردند، دچار شکم درد، مشکلات کلیوی و خونی شدند.آلودگی طولانی مدت و مداوم با نیکل کربونیل با افزایش شیوع سرطان ریه و سینوس ها همراه است . محصولات حاصل از تجزیهء نیکل (نیکل اکسید و کربن مونوکسید)، نسبت به خود نیکل کربونیل سمیت کمتری دارند. در موشهایی که برای مدتی ترکیبات نیکل را استنشاق کرده بودند، ترکیباتی از نیکل که به سختی در آب حل می شوند، موجب سرطان شدند و ترکیباتی که در آب حل می شدند، موردی را ایجاد نکردند.بخش سلامت و سرویسهای انسانی (DHHS)، نیکل و ترکیبات خاصی از آن را بعنوان عوامل سرطانزای احتمالی معرفی کرده اند. در کارگران پالایشگاهها و کارخانجات آبکاری که غلظتهای بالایی از ترکیبات نیکل را استنشاق کرده بودند، سرطان ریه و سینوسهای بینی مشاهده شده بود. IARC ، نیکل و ترکیباتش را در گروه ۲B (عوامل سرطانزای احتمالی) طبقه بندی کرده اند.
طبقه بندی مواد شیمیایی
ماده ، به هر چیزی که حجمی را اشغال کند و جرمی داشته باشد، اطلاق میشود. مواد شیمیایی به موادی اطلاق میگردد که معمولا از طریق سنتز شیمیایی تهیه میشوند و یا اینکه منشأ طبیعی داشته و مواد اولیه تهیه سایر مواد شیمیایی به حساب میآیند.
طبقه بندی مواد شیمیایی
مواد شیمیایی بطور عمده به دو گروه بزرگ مواد معدنی و مواد آلی تقسیم بندی میشوند. هر یک از این دو گروه ، در دو مبحث شیمی آلی و شیمی معدنی بررسی میشوند. در این مطالعه ، خواص فیزیکی و شیمیایی مواد آلی و معدنی ، منابع ، طریقه سنتز و واکنشها و ... مورد بررسی قرار میگیرند.
مواد شیمیایی آلی
در قدیم ، ماده آلی به مادهای اطلاق میگردید که بوسیله بدن موجودات زنده ساخته میشد. تا اینکه در سال 1828 ، "وهلر" (Wohler) دانشمند آلمانی ، برای اولین بار جسمی به نام اوره به فرمول CO(NH2)2 را در آزمایشگاه از یک ترکیب معدنی به نام ایزوسیانات تهیه نمود و از آن پس معلوم شد که میتوان مواد آلی را نیز در آزمایشگاه ساخت.
امروزه بیش از یک میلیون نوع ماده آلی شناخته شده است که بسیاری از آنها را در آزمایشگاهها تهیه میکنند. مواد آلی ، به مواد غیر معدنی گفته میشود و با مواد معدنی تفاوتهای کلی در چند مورد دارند.
مواد شیمیایی معدنی
اگر شیمی آلی به عنوان شیمی ترکیبات کربن ، عمدتا آنهایی که شامل هیدروژن یا هالوژنها به علاوه عناصر دیگر هستند، تعریف شود، شیمی معدنی را میتوان بطور کلی به عنوان شیمی عناصر دیگر در نظر گرفت که شامل همه عناصر باقیمانده در جدول تناوبی و همینطور کربن ، که نقش عمدهای در بیشتر ترکیبات معدنی دارد، میگردد.
شیمی آلی - فلزی ، زمینه وسیعی که با سرعت زیاد رشد میکند، به علت اینکه ترکیبات شامل پیوندهای مستقیم فلز - کربن را بررسی میکند دو شاخه را بهم مرتبط میسازد. همانطوری که میتوان حدس زد، قلمرو شیمی معدنی با فراهم کردن زمینههای تحقیقی اساسا نامحدود ، بسیار گسترده است.
مقایسه مواد آلی و مواد معدنی
مواد شیمیایی آلی و معدنی با همدیگر تفاوتهای کلی دارند که عبارتند از:
- در تمام مواد آلی حتما کربن وجود دارد، در صورتی که مواد معدنی بدون کربن بسیارند. ضمنا در ترکیبات آلی ، اتمهای کربن میتوانند با یکدیگر ترکیب شوند و زنجیرهای طویل تشکیل دهند، در حالیکه این خاصیت در عناصر دیگر خیلی کمتر دیده میشود.
- مقاومت مواد آلی در برابر حرارت از مواد معدنی کمتر است.
- اغلب واکنشهای میان مواد آلی کند و دو جانبه یا تعادلی هستند، در صورتیکه اغلب واکنشهای معدنی تند میباشند.
- در ترکیبات آلی ، ممکن است 2 یا چند جسم مختلف با فرمولهای ساختمانی مختلف ، دارای یک فرمول مولکولی باشند که در این صورت به آنها ایزومر یا همفرمول گفته میشود. مثلا الکل معمولی C2H5OH با جسمی به نام اتر اکسید متیل CH3OCH3 همفرمول یا ایزومر است. زیرا هر دو دارای فرمول بسته یا مولکولی C2H6O هستند، در صورتی که پدیده ایزومری در ترکیبات معدنی وجود ندارد.
تقسیم بندی مواد شیمیایی آلی
عناصر تشکیل دهنده ترکیبات شیمیایی آلی به ترتیب فراوانی مطابق زیر است:
فلزات , هالوژنها , C , H , O , N , S , P , As . فراوانترین چهار عنصر N , O , H , C عناصر اصلی سازنده مواد آلی به حساب میآیند. زیرا اغلب اجسام آلی از این چهار عنصر تشکیل یافتهاند و با توجه به همین مطلب ، مواد آلی را به چهار دسته کلی تقسیم میکنیم:
هیدروکربنهای ساده
ترکیباتی هستند که فقط از H , C درست شدهاند و به همین دلیل ، هیدروکربن شدهاند. آنها با فرمول کلی CxHy نمایش میدهند. بسته به اینکه y , x چه اعدادی باشند، هیدروکربنهای گوناگون یافت میشوند.
هیدروکربنهای اکسیژندار
ترکیباتی هستند که از O , H , C درست شده اند و با فرمول کلی CxHyOz نشان داده میشوند.
هیدروکربنهای نیتروژندار
ترکیباتی هستند که از N , H , C درست شدهاند و با فرمول کلی CxHyNt نشان داده میشوند.
هیدروکربنهای اکسیژن و نیتروژن دار
ترکیباتی هستند که علاوه بر H ، C ، اکسیژن و نیتروژن و با فرمول کلی CxHyOzNt نمایش داده میشوند.
تعیین دمای ذوب
تعیین دمای ذوب:
دمای ذوب را عمدتا به دو طریق زیر تعیین میکنند:
1-لوله تیل
2-دستگاههای اندازه گیری دقیق میکروسکوپی
1- لوله تیل:
وسیله ساده ای است که به سهولت قابل دسترسی است.
لوله تیل به نحوی طراحی شده است که وقتی در آن روغن می ریزیم و لوله را گرم می کنیم، در آن تبادل گرمایی صورت می گیرد.
به نحوی که توزیع دما در سراسر روغن داخل لوله یکنواخت می شود. چنانچه لوله تیل در دسترس نباشد از یک بشر کوچک 50 یا 100 میلی لیتری می توان به عنوان حمام استفاده کرد.
آماده کردن نمونه:
مقدار کمی از ترکیب جامد را در هاون بسایید و به صورت پودر نرمی در آورید. یک لوله مویین به طول حدود 10 سانتیمتر بردارید و یک انتهای آن را با استفاده از شعله مسدود کنید. انتهای باز لوله را در توده نرم شده فرو کنید تا مقداری از آن داخل لوله شود. سپس ته لوله را چند بار آهسته روی میز بزنید تا تمام پودر در انتهای آن قرار گیرد. همچنین می توانید یک لوله شیشه ای را که ابتدا و انتهای آن باز است به طور عمودی روی میز قرار دهید و لوله مویین را از سمت انتهای بسته در آن رها کنید. لوله مویین را به کمک یک نخ یا کش به دماسنج متصل کنید به طوری که انتهای لوله مویین و بخش جیوه ای دماسنج هم تراز شوند. اکنون دماسنج و لوله مویین را به کمک پایه و گیره در داخل حمام روغن قرار دهید. حمام را به آهستگی با شعله (چراغ بنسن) گرم کنید و دمای ابتدا و انتهای ذوب شدن را از روی درجات دماسنج با دقت بخوانید و یادداشت کنید. اگر دمای ذوب یک ترکیب شناخته شده نیست معمولا دو لوله مویین حاوی ترکیب آماده می کنند. با لوله مویین اول نقطه ذوب را سریعا اندازه می گیرند. سپس دمای حمام را تا حدود 30 درجه کاهش می دهند و با استفاده از لوله مویین دوم نقطه ذوب را به آرامی و با دقت تعیین می کنند. اگر لوله مویین در دسترس نباشد با استفاده از یک لوله شیشه ای به قطر حدود 0.5 سانتی متر و طول 25 سانتی متر لوله مویین بسازید.
نقاط ذوب مخلوط ها:
دمای ذوب هر ماده بلوری خالص، یک خاصیت فیزیکی آن ماده است و می توان از آن برای شناسایی یک ترکیب استفاده کرد. به طور کلی افزایش تدریجی و پی در پی ناخالصی به یک ماده خالص سبب می شود که به نسبت مقدار ناخالصی افزوده شده نقطه ذوب کاهش یابد.
دمای تقطیر و جوش:
فشار بخار مایعات، براثر گرم شدن آنها زیاد می شود تا حدی که فشار بخار مایع برابر فشار هوا می شود. در این حالت جوشیدن مایع قابل رویت است. این دما را نقطه جوش یا دمای جوش می نامند. با کاهش فشار، نقطه جوش نیز پایین می آید زیرا انرژی گرمایی کمتری برای برقراری تعادل بین فشار بخار مایع و فشار هوا (که کم شده است) لازم است. نقطه جوش در فشار یک اتمسفر را نقطه جوش عادی (نرمال) می گویند. فرایند تبخیر و سپس میعان مجدد یک مایع را تقطیر می گویند. این روش برای جدا کردن مخلوط چند جزء که نقاط جوش متفاوتی دارند سودمند است. همچنین یک روش اساسی برای تخلیص مایعات به شمار می آید. نقطه جوش مایع خالصی که در طول عمل تقطیر تجزیه نمی شود، دقیق و در تمام مدت جوش ثابت است. تعیین نقطه جوش (bp) با دو روش به آسانی امکان پذیر است. استفاده از این دو روش به مقدار ماده موجود بستگی دارد.
۱. چنانچه مایع به مقدار کافی یا زیاد در دسترس باشد، نقطه جوش آن را می توان به روش تقطیر ساده و به کمک دماسنج تعیین کرد.
۲. در صورتی که مقدار مایع کم باشد، از روش نقطه جوش میکرو استفاده می شود.
تعیین نقطه جوش به روش میکرو:
در این روش از لوله آزمایشی به قطر داخلی 5 میلی متر و طول تقریبی 12 سانتیمتر استفاده می شود. مقداری از مایع مورد نظر (0.2 تا 0.5 میلی لیتر) را به وسیله پی پت یا قطره چکان به درون لول آزمایش می ریزیم. سپس لوله مویینی را که یک انتهای آن مسدود شده است به طور واژگون از انتهای باز آن به درون لوله می اندازیم. بعد این لوله را به وسیله نخ یا نوار لاستیکی به دماسنج می بندیم. همانگونه که در تعیین نقطه ذوب عمل کردیم. انتهای لوله و دماسنج باید در یک سطح باشند. این مجموعه را در حمام روغن قرار می دهیم و به آرامی گرم می کنیم. پس از مدتی گرم کردن، جریان منظم و یکنواختی از حباب هوا از انتهای لوله مویین خارج می شود. در این مرحله گرما را قطع می کنیم و ملاحظه می شود که جریان حباب هوا قطع می شود و سپس مقداری از مایع وارد لوله مویین می شود. در این لحظه عدد دماسنج را می خوانیم و ثبت می کنیم. این دما، نقطه جوش مایع است. در تعیین نقطه جوش به روش میکرو مشکلاتی به شرح زیر پیش می آید:
*چون مقدار مایع اندک است، در صورت افزایش ناگهانی گرما احتمال بخار شدن آن وجود دارد، و یا اینکه ممکن است نقطه جوش به دست آمده بیشتر از مقدار واقعی باشد.
* اگر گرم کردن کافی نباشد، در نزدیکی نقطه جوش، در صورت گرما، ممکن است مایع از لوله آزمایش، وارد لوله مویین شود، زیرا در این لحظه فشار بخار مایع پایینتر از فشار هواست. نقطه جوش به دست آمده در این روش به علت تجربه ناکافی آزمایش کننده و خطای چشم، تقریبی، و غالبا کمتر از مقدار واقعی است.
با تشکر از دوست عزیزم جناب آقای فراهانی
کاتالیزور
کاتالیزور ماده ای است که سرعت یک واکنش شیمیایی را افزایش می دهد بدون آنکه خود در جریان واکنش مصرف شود.
ریشه لغوی
کاتالیزور از دو صفت کاتا و لیزور تشکیل شده است. در زبان یونانی "کاتا" به معنای پائین ، افتادن ، یا پائین افتادن است و "لیزور" به معنی قطعه قطعه کردن میباشد. در برخی زبانها کاتالیزور را به معنی گردهم آوردن اجسام دور از هم معرفی کرده اند.
تاریخچه
اولین گزارش استفاده از کاتالیزور ، مربوط به کریشف میباشد که با استفاده از یک اسید به عنوان کاتالیزور توانست نشاسته را به قند ، هیدرولیزکند. بعدها دیوی توانست واکنش اکسیداسیون هیدروژن را با اکسیژن در حضور کاتالیزورپلاتین انجام دهد که این واکنش یک واکنش گرما گیر است و در نتیجه هنگام انجام واکنش جرقه تولید میشد.
اولین کار در توضیح اینکه چرا یک واکنش کاتالیزوری انجام میگیرد و کاتالیزور چه نقشی دارد، توسط "فارادی" انجام شد. بیشترین بهرهبرداری از کاتالیزور در جنگ جهانی بود.
انقلاب تکنولوژی اصلی در زمینه کاتالیزور مربوط به نیمه دوم قرن 20 یعنی بین سالهای1980 ـ 1950 میباشد.دهه 1960 ـ 1950 دهه ای است که با تولید کاتالیزورهای زیگر _ ناتا ترکیبات بسیار مهم و استراتژیک ساخته شد.
انواع کاتالیزور
کاتالیزور به دو نوع کاتالیزور مرغوب و نامرغوب تقسیم میشود:
- کاتالیزور مرغوب: کاتالیزور مرغوب به کاتالیزوری گفته میشود که فقط اجازه تشکیل یک نوع محصول را بدهد.
- کاتالیزور نامرغوب: اگر در حضور کاتالیزور محصولات متفاوتی امکان تشکیل داشته باشند کاتالیزور نامرغوب تلقی میشود.
چگونگی عمل کاتالیزور
تجربه نشان داده است که واکنش با کاتالیزور در دمای کمتری صورت میگیرد و همچنین کاتالیزور ، انرژی اکتیواسیون را پائین میآورد یا کاهش میدهد یا باعث میشود مولکولهای درشت به مولکولهای کوچکتر ، قطعهقطعه یا شکسته شوند.
کاتالیزور واکنش را میتوان بدون تغییر در پایان واکنش بدست آورد. مثلا سرعت تجزیه KClO3 را با مقدار کمی MNO2 میتوان فوقالعاده زیاد کرد. در معادلهای که برای این تغییر نوشته میشود ، کاتالیزور را بالای پیکان میگذارند ، زیرا کاربرد آن در استوکیومتری کل واکنش اثری ندارد:
مکانیسم واکنش کاتالیزوردار
کاتالیزور نمیتواند موجب وقوع واکنشهایی شود که از نظر ترمودینامیک امکان وقوع ندارند. بعلاوه صرفا حضور کاتالیزور نیست که (احتمالا بعنوان یک بخش فعالکننده) موجب اثر بر سرعت واکنش میشود. در یک واکنش کاتالیزوردار ، کاتالیزور در یک مرحله عملا مصرف میشود و در مرحله بعدی بار دیگر تولید میگردد و این عمل بارها تکرار میگردد، بدون آنکه کاتالیزور دچار تغییر دائمی شود.
بنابراین کار کاتالیزور آن است که راه تازه ای برای پیشرفت واکنش میگشاید. بدین ترتیب مکانیسم کاتالیزوردار با یک واکنش بیکاتالیزور تفاوت دارد. انرژی فعال سازی راهی که واکنش به کمک کاتالیزور طی میکند، کمتر از انرژی فعالسازی راهی است که همان واکنش بدون کاتالیزور میپیماید (شکل 1) 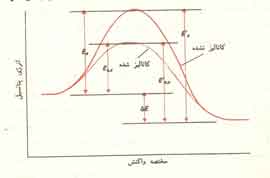
این واقعیتی است که علت سریعتر شدن واکنش را توجیه میکند. وقتی کاتالیزور بکار برده میشود، مولکولهای نسبتا بیشتری انرژی لازم برای یک برخورد موفق پیدا میکنند (شکل 2). بدین ترتیب عده کل برخوردهای موثر در واحد زمان، که موجب انجام واکنش میشوند، افزایش مییابد.
در شکل 1 به دو نکته دیگر نیز پی میبریم. نخست آنکه تغییرات انرژی برای واکنش کاتالیزوردار و واکنش بیکاتالیزور یکسان است. دیگر آنکه انرژی فعال سازی واکنش معکوس نیز به هنگام استفاده از کاتالیزور کاهش مییابد و مقدار کاهش آن درست برابر کم شدن انرژی فعال سازی واکنش کاتالیزوردار اصلی است. این بدان معنی است که کاتالیزور بر یک واکنشی و واکنش معکوس آن اثر یکسان دارد. اگر یک کاتالیزور سرعت یک واکنش را دو برابر کند، همان کاتالیزور سرعت واکنش معکوس آن را نیز دو برابر خواهد کرد.
کاتالیزورهای طبیعی (آنزیم)
بسیاری از فرایندهای صنعتی به اعمالی بستگی دارند که با کاتالیزور صورت میگیرند. ولی کاتالیزورهایی که برای انسان مورد اهمیت بیشتری دارند، کاتالیزورهای طبیعی یعنی آنزیمها هستند. این مواد فوق العاده پیچیده ، فرایندهای حیاتی مانند گوارش و سنتز سلولی را کاتالیز میکنند.
عده زیادی از واکنشهای شیمیایی پیچیده که در بدن صورت میگیرد و برای حیات ما ضرورت دارد، به علت اثر آنزیمها در دمای پائین بدن امکان وقوع پیدا میکنند. هزاران آنزیم وجود دارند که هر یک وظیفه خاصی را انجام میدهند. تحقیق درباره ساختمان و عمل آنزیمها ، نویدهای فراوانی درباره پیشرفت شناخت عامل بیماری و مکانیسم رشد میدهد. 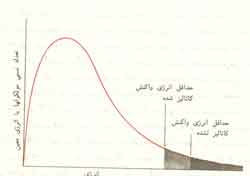
کاتالیزور همگن و ناهمگن
در کاتالیزور همگنماده ای که بعنوان کاتالیزور کار میکند، با مواد واکنشدهنده در یک فازند، ولی در یک کاتالیزور ناهمگن یا کاتالیزور سطحی ، مواد واکنشدهنده و کاتالیزور در دو فاز مجزا کنار هم هستند و واکنش در سطح کاتالیزور صورت میگیرد.
کاتالیزور همگن
نمونه ای از کاتالیزور همگن در فاز گازی ، اثر کلر در تجزیه دینیترون اکسید است. گاز دینیترون اکسید ، در دمای اتاق ، گاز نسبتا بیاثری است، اما در دماهای نزدیک به صد درجه طبق معادله زیر تجزیه می شود.
مطالعات سینتیک نشان میدهد که واکنش مذکور بر اثر برخورد بین دو ملکول کلر کاتالیز میشود.
کاتالیزور همگن در محلول نیز ممکن است صورت گیرد. بسیاری از واکنشها بوسیله اسیدها و بازها کاتالیز میشوند. تجزیه هیدروژن پراکسید در حضور پون یدید کاتالیز میشود.
کاتالیزور ناهمگن
کاتالیزور ناهمگن عمدتا از طریق جذب سطحی شیمیایی مواد واکنش دهنده بر سطح کاتالیزور صورت میگیرد. جذب سطحی فرآیندی است که در جریان آن مولکولها به سطح جسمی جامد میچسبند. مثلا در ماسکهای گازی ، زغال به عنوان یک ماده جاذب برای گازهای زیان آور بکار میرود.
در جذب سطحی فیزیکی معمولی ، مولکولها ، بوسیله نیروهای و اندروالسی به سطح ماده جاذب ، گیر میکنند. بنابراین مولکولهایی از گاز که جذب سطحی شدهاند، تا همان حد تحت تاثیر قرار گرفتهاند که گویی مایع شده باشند.
در جذب سطحی شیمیایی ، مولکولهای جذب شده ، با پیوندهایی که قابل مقایسه با پیوندهای شیمیایی است، به سطح ماده کاتالیزور نگه داشته میشوند. در فرایند تشکیل پیوند با ماده جاذب ، مولکولهایی که بطور شیمیایی جذب شدهاند، دچار تغییر آرایش الکترونی درونی میشوند. پیوندهای درون بعضی از مولکولهای کشیده و ضعیف و حتی پیوند بعضی از آنها شکسته میشوند.
مثلا هیدروژن بصورت اتمی بر سطح پلاتین جذب میشود. بنابراین تعدادی از ملکولها که بطور شیمیایی جذب سطحی شدهاند، به صورت کمپلکس فعال شده یک واکنشی که در سطح کاتالیزور شده، عمل میکند.
مکانیسم جذب سطحی شیمیایی:
تاکنون مکانیسم دقیق جذب سطحی شیمیایی و کاتالیز سطح کاملا فهمیده نشده است، فقط فرضهایی قابل قبول برای مکانیسم چند واکنش خاصی مطرح شده است:
- نظری دال بر اینکه نقصها یا بینظمیهای شبکه در سطح کاتالیزور ، جای فعالی برای عمل کاتالیزور است، اولین فرضیه برای توضیح عمل تقویت کنندههای کاتالیزورهای مناسب است. تقویت کننده ها موادی هستند که فعالیت کاتالیزور ها را زیاد میکنند. مثلا در سنتز آمونیاک
اگر کاتالیزورآهن با مقدار کمی پتاسیم یا وانادیم آمیخته شده باشد، بیشتر موثر واقع میشود.
تبلور
انتخاب حلال مناسب نکته اساسی و مهم در عمل تبلور محسوب می شود. حلال مناسب حلالی است که در دمای معمولی جسم را به مقدار جزئی در خود حل کند، ولی در گرما و به ویژه در دمای جوش، این انحلال به آسانی صورت گیرد.
عامل دیگر در انتخاب حلال مناسب، توجه به قطبیت آن است که با توجه به ساختمان ماده مورد نظر انتخاب می شود.
زیرا ترکیبات قطبی در حلالهای قطبی و ترکیبات غیر قطبی در حلالهای غیر قطبی حل می شوند.
به هنگام انتخاب حلال مناسب برای تبلور، به نکات زیر توجه کنید:
حلال در دمای معمولی (دمای آزمایشگاه) نباید ترکیب را حل کند، اما در نقطه جوش خود باید حداکثر ترکیب یا تمام آن را در خود حل کند.
نقطه جوش حلال نباید از نقطه ذوب ترکیب مورد نظر بیشتر باشد. زیرا در این صورت، پیش از اینکه دمای حلال به نقطه جوش آن برسد، جسم در حلال ذوب می شود. (در پدیده تبلور، جسم باید در حلال حل شود).
حلال و جسم حل شده نباید با هم واکنش بدهند.
تا حد امکان نقطه جوش حلال پایین باشد تا به آسانی تبخیر شود.
مراحل متوالی زیر، پس از انحلال جسم جامد در حلال باید اجرا شود:
چنانچه محلول به شدت رنگی و یا ناخالص باشد، گرم کردن را قطع کنید پس از اینکه محلول، اندکی خنک شد، کمی پودر زغال به آن اضافه کنید. زغال به دلیل دارا بودن سطح فعال زیاد می تواند ناخالصیها و رنگ را به خود جذب کند. سپس مجددا محلول را گرم کنید.
به منظور جداسازی ناخالصیهای نامحلول، محلول را گرم و صاف کنید.
برای اینکه بلورها پدیدار شوند محلول صاف شده را به تدریج سرد کنید.
بلورها را به وسیله صاف کردن جدا کنید.
بلورها را با مقدرا کمی حلال سرد بشویید.
بلورها را خشک کنید.
در تبلور، برای ظاهر شدن از چند روش استفاده می شود:
یک میله شیشه ای (همزن) را از سطح مایع و کنار آن به جدار ظرف بسایید(خراش دهید). حرکت میله باید به صورت عمودی و سریع باشد. به نظر می رسد که با عمل سایش، به مولکولهای جسم انرژی داده می شود و این انرژی باعث نزدیک شدن مولکولها به یکدیگر می شود و به این ترتیب، تشکیل هسته اولیه آسان می شود.
ظرف حاوی محلول را به وسیله قراردادن آن در حمام آب و یخ یا در یخچال سرد کنید.
یک تکه از بلور ترکیب را به عنوان هسته اولیه در ارلن مایر بیندازید این عمل را بذرافشانی و تکه بلور را بذر می نامند.
کاتالیست های عمل آوری شده با هیدروژن
کاتالیست های عمل آوری شده با هیدروژن
با وجود این چنانچه حذف نیتروژن حائز اهمیت باشد کاتالیست هایی که از ترکیبات نیکل – کبالت – مولیبدن یا نیکل – مولیبدن آلومین پایه باشند. از کارایی بیشتری برخوردارند. معمولا حذف نیتروژن از جریان های هیدروکربنی دشوارتر از حذف گوگرد است و هر عملی که بتواند مقدار اضافی نیتروژن را به حد مطلوب کاهش دهد قادر خواهد بود به طرز موثرتری مقدار اضافی گوگرد را حذف کند. معمولا کاتالیست های حاوی نیکل را پیش از رساندن به دمای واکنش فعال می کنند. این عمل به روش پیش سولفیدی کردن آن ها با کربن دی سولفید مرکاپتان یا دی متیل سولفید انجام می شود. ولی بعضی پالاشگرها این کاتالیست را از طریق تزریق مواد شیمیایی سولفید کننده در خوراک نفتی به هنگام راه اندازی فعال می کنند. واکنش سولفیدی شدن به شدت گرما زاست و باید دقت کرد که از دماهای بالا در طول فعالسازی اجتناب شود.
کاتالیست های کبالت – مولیبدن برای گوگردزدایی و کاتالیست های نیکل – مولیبدن برای نیتروژن زدایی مناسب هستند ولی هر دو کاتالیست می توانند گوگرد و نیتروژن را جدا کنند. کاتالیست های نیکل و مولیبدن برای هیدروژن دار کردن فعالتر از کاتالیست های کبالت – مولیبدن می باشند. که در شرایط عملیاتی مشابه بیشتر به سیر کردن حلقه های آروماتیکی می پردازند. به طور خلاصه اگر هدف کاهش گوگرد کاتالیست های کبالت – مولیبدن در مقایسه با کاتالیست های نیکل مولیبدن می توانند شرایط عملیاتی ملایمتر و با مصرف هیدروژن کمتر مقدار گوگرد را تا حد مورد نظر کاهش دهد. اگر کاهش نیتروژن یا سیر کردن حلقه های آروماتیکی در مد نظر باشد کاتالیست نیکل مولیبدن ترجیح داده می شود.
شناسایی عناصر در ترکیبات آلی (ذوب قلیایی)
شناسایی عناصر در ترکیبات آلی (تجزیه کیفی مواد آلی به روش ذوب قلیایی جهت تشخیص ازت، گوگرد و هالوژنها)
1- مقدمه
برای تشخیص عناصر موجود در ترکیبات آلی (عناصر موجود در ترکیبات آلی معمولی عبارتند از: کربن، هیدروژن، اکسیژن، نیتروژن، گوگرد و هالوژنها) ابتدا باید آنها را به ترکیبات معدنی یونیزه که قابل شناسایی باشد تبدیل کرد.
این تبدیل ممکن است به روشهای مختلف صورت گیرد ولی بهترین روش ذوب ترکیبات با فلز سدیم است. در این روش سیانید سدیم (NaCN)، سولفید سدیم (Na2S) و هالید سدیم (NaX) تشکیل میشود که به آسانی قابل تشخیص هستند.
در شکل ماده (1) ترکیب آلی است
معمولا سدیم به مقدار اضافی به کار برده میشود. در غیر اینصورت اگر گوگرد و نیتروژن هردو وجود داشته باشند. احتمالا تیوسیانات سدیم (NaSCN) تشکیل میشود. در این صورت در تشخیص نیتروژن به جای آبی پروس رنگ قرمز مشاهده میشود زیرا بجای یون (CN-)، یون (SCN-) خواهیم داشت. اما با سدیم اضافی تیوسیانات تشکیل شده تجزیه میشود و جواب درست به دست می آید.
به مخلوط حاصل آب اضافه کرده مخلوط قلیایی را صاف نموده و سپس به آن (FeSO4) اضافه کنید در این صورت فروسیانید سدیم تشکیل میشود.
وقتی محلولهای قلیایی نمکهای فروی بالا جوشانده میشود بر اثر اکسیژن هوا کمی یون فریک تشکیل میشود. (بر اثر سولفوریک اسید رقیق هیدروکسیدهای فرو و فریک تشکیل شده حل میشوند) فروسیانیدها با نمک فریک تشکیل فروسیانید فریک (آبی پروس) میدهند.
برای اسیدی کردن محیط نباید از (HCl) استفاده کرد زیرا به علت تشکیل (FeCl6) رنگ زرد در محیط ایجاد میشود و به جای آبی پروس رنگ سبز ظاهر میشود. به همین دلیل کلرید فریک نیز نباید اضافه شود. همانطوری که قبلا ذکر شده است بر اثر اکسیداسیون به وسیله هوا در محیطهای قلیایی گرم به مقدار کافی یونهای فریک تشکیل میشود بنابراین نیازی به افزایش یون فریک نیست، افزایش مقدار کمی محلول رقیق فلئورید پتاسیم ممکن است به تشکیل آبی پروس در محلول که به آسانی قابل صاف شدن است کمک نماید (Fe3+ با F- تولید FeF63- میکند که پایدار است و باعث خارج شدن Fe3+ از محیط عمل میشود).
گوگرد به صورت یون سولفید را میتوان به وسیله استات سرب و استیک اسید و یا به و سیله پلمبیت سدیم (محلول قلیایی استات سرب) به صورت رسوب سولفید سرب (PbS) سیاه رنگ تشخیص داد.
رسوب سیاه رنگ
برای تشخیص یونهای هالوژن (Cl, Br, I) از اثر محلول نیترات نقره در محیط اسید نیتریکی استفاده میشود در این صورت هالید نقره به صورت رسوب حاصل میشود.
کروماتوگرافی لایه نازک (TLC)
کروماتوگرافی لایه نازک (TLC)
کروماتوگرافی لایه نازک نوعی کروماتوگرافی جذبی جامد – مایع است و اصول آن مانند کروماتوگرافی ستونی است. ولی در این مورد جسم جاذب جامد را به صورت یک لایه نازک در روی یک قطعه شیشه یا پلاستیک محکم پخش میکنند. یک قطره از محلول نمونه یا مجهول را در نزدیکی لبه صفحه میگذارند و صفحه را همراه مقدار کافی از حلال استخراج کننده در ظرفی قرار میدهند. مقدار حلال باید آنقدر باشد که فقط به سطح زیر لکه برسد (شکل الف). حلال به طرف بالای صفحه میرود و اجزاء مخلوط را با سرعتهای متفاوت با خود میبرد. در نتیجه ممکن است تعدادی لکه روی صفحه ظاهر شود. این لکه ها روی یک خط عمود بر سطح حلال ظرف قرار میگیرند (شکل ب).
این روش کروماتوگرافی بسیار آسان است و به سرعت هم انجام میشود. این روش برای تفکیک اجزاء یک مخلوط بسیار مفید است و همچنینی میتوان از آن برای تعیین بهترین حلال استخراج کننده جهت کروماتوگرافی ستونی استفاده کرد.
در TLC میتوان از همان مواد جامد که در کروماتوگرافی ستونی استفاده میشود استفاده کرد و در این میان سیلیکا و آلومینا بیشتر به کار میرود. معمولا جسم جاذب را با مقدار کمی از ماده نگهدارنده مانند گچ شکسته بندی، کلسیم سولفات و یا نشاسته مخلوط میکنند تا جسم جاذب چسبندگی لازم را پیدا کند و به صفحه بچسبد. صفحه ها را میتوان قبل از مصرف تهیه کرد و یا از ورقه های پلاستیکی آماده که در بازار موجود است استفاده نمود.
یکی از مزایای مشخص TLC آن است که احتیاج به مقدار بسیار کمی از نمونه دارد. در بعضی موار میتوان تا مقدار 9-10 گرم را تشخیص داد. اما ممکن است اندازه نمونه تا 500 میکرو گرم برسد. در نمونه های زیاد میتوان از تجربه های تهیه ای استفاده کرد. در این تجربه ها لکه های مختلف را میتراشند و با یک حلال مناسب میشویند (استخراج میکنند). و برای شناسایی (از طریق طیف سنجی) به کار میبرند.
تشخیص لکه های رنگین در روی کروماتوگرام آسان است و برای تعیین محل لکه های اجسام بیرنگ روشهای متعددی وجود دارد. برای مثال میتوان با تابش نور ماوراء بنفش به صفحه محل لکه، ترکیبهایی را که خاصیت فلوئورسانس دارند مشخص کرد. به روش دیگر میتوان جسم جاذب را با ماده فلوئورسانس دار بی اثر دیگری مخلوط کرد. هنگامی که نور ماوراء بنفش به این صفحه بتابد، لکه اجسامی که نور ماورای بنفش را جذب میکنند ولی خاصیت فلوئورسانس ندارند در زمینه فلورسانس دار صفحه به صورت تیره رنگ ظاهر میشوند. در بسیاری موارد دیگر، از معرفهای آشکارساز دیگری استفاده میکنند. این معرفها را میتوان بر روی کروماتوگرام پاشید و لکه ها را ظاهر کرد. سولفوریک اسید، که بسیاری از ترکیبات آلی را به ذغال تبدیل میکند و محلول پتاسیم پرمنگنات نمونه هایی از معرفهای آشکار ساز هستند که به این روش مصرف میشوند. ید نیز معرف آشکار ساز دیگری است که مصرف میشود. در این مورد صفحه را دز ظرفی میگذارند که محیط آن از بخار ید اشباع باشد. بسیاری از ترکیبات آلی ید را جذب میکنند و لکه آنها روی کروماتوگرام رنگین (معمولا قهوه ای) میشود.
در شرایط معین سرعت حرکت ترکیب نسبت به سرعت پیشرفت حلال (Rf) خاصیت مشخصی از ترکیب است. برای تعیین این مقدار مسافتی را که جسم از خط شروع تا وسط لکه را طی کرده است اندازه میگیرند و آنرا به مسافتی که حلال پیموده تقسیم میکنند. این مسافت را با خط شروع یکسانی میسنجند.
بخش عملی
تفکیک مواد رنگی برگ سبز
چند میلی لیتر از مخلوط 2 به یک اتر نفت و اتانول را همراه با چند برگ سبز در هاونی بگذارید و برگها را با دسته هاون له کنید. مایع بدست آمده را به یک قیف جدا کننده منتقل کنید و همان حجم آب مقطر به آن اضافه کنید و تکان دهید. فاز آبی پایینی را دور بریزید. این شستشو را دو بار انجام دهید و هر بار فاز آبی را دور بریزید. و آب تازه اضافه کنید.لایه آلی (بالایی) را به ارلن کوچکی منتقل کنید و به آن 2 گرم سدیم سولفات بدون آب اضافه کنید (برای آب گیری).
یک نوار 10 سانتی از ورقه کروماتوگرام سیلیکاژل تهیه کنید و یک لکه 1 الی 2 میلی متری از محلول ماده رنگی را طوری بر روی صفحه قرار دهید که حدود 1 و نیم سانتی متر از انتهای آن فاصله داشته باشد (برای گذاشتن لکه از لوله مویین تمیز استفاده کنید). صبر کنید تا لکه خشک شود. برای جداسازی از حلال بنزن – استون با نسبت 7 – 3 (حجمی) مطابق توضیحات بالا استفاده کنید.
ممکن است تا هشت لکه رنگین مشاهده شود. این لکه ها به ترتیب کاهش مقدار Rf عبارتند از کاروتنها (دو لکه نارنجی)، کلروفیل a (آبی – سبز)، کلروفیل b (سبز) و زانتوفیلها (چهار لکه زرد).
رنگ شعله

رنگ شعله
رنگ | ماده شیمایی |
Carmine | لیتیوم کلرید |
قرمز | استرنسیوم کلرید |
نارنجی | کلسیم کلرید |
زرد | سدیم کلرید |
زرد مایل به سبز | بوراکس |
سبز | مس سولفات |
آبی | مس کلرید |
بنفش | 3 parts Potassium Sulfate |
ارغوانی | پتاسیم کلرید |
سفید | منیزیوم سولفات |
Flame Colorants
| Color | Chemical |
| Carmine | Lithium Chloride |
| Red | Strontium Chloride |
| Orange | Calcium Chloride (a bleaching powder) |
| Yellow | Sodium Chloride (table salt) or Sodium Carbonate |
| Yellowish Green | Borax |
| Green | Copper Sulfate |
| Blue | Copper Chloride |
| Violet | 3 parts Potassium Sulfate 1 part Potassium Nitrate (saltpeter) |
| Purple | Potassium Chloride |
| White | Magnesium Sulfate (Epsom salts) |
| Color | Compound |
|---|---|
| Red | strontium salts, lithium salts lithium carbonate, Li2CO3 = red strontium carbonate, SrCO3 = bright red |
| Orange | calcium salts calcium chloride, CaCl2 calcium sulfate, CaSO4·xH2O, where x = 0,2,3,5 |
| Gold | incandescence of iron (with carbon), charcoal, or lampblack |
| Yellow | sodium compounds sodium nitrate, NaNO3 cryolite, Na3AlF6 |
| Electric White | white-hot metal, such as magnesium or aluminum barium oxide, BaO |
| Green | barium compounds + chlorine producer barium chloride, BaCl+ = bright green |
| Blue | copper compounds + chlorine producer copper acetoarsenite (Paris Green), Cu3As2O3Cu(C2H3O2)2 = blue copper (I) chloride, CuCl = turquoise blue |
| Purple | mixture of strontium (red) and copper (blue) compounds |
| Silver | burning aluminum, titanium, or magnesium powder or flakes |
با تشکر از سرکار خانم مهندس ترکاشوند
Raman Spectroscopy
مقدمه:
طیف نمایی رامان چه در اصول و چه در عمل تا اندازهای از طیف نمایی جذبی و گسیلی متفاوت است. اما آنچنان وسیله مهمی در تحلیل مولکولی است که بایستی در اینجا بطور خلاصه ذکر میکنیم. اگر گاز یا مایعی تحت تابش یک خط قوی با فرکانس اختیاری (v0 به عنوان مثال ، یکی از خطوط صادره از لامپ جیوه) قرار گیرد، نور پراکنده شده عمدتا دارای همان فرکانس است.
اما یک یا چند خط ضعیف جابجایی ممکن است در طرف طول موجهای بلند مثلا در فرکانسهایνs ظاهر شوند که اینها به خطوط استوکس معروفند. حتی در طرف طول موجهای کوتاه ممکن است خطوط ضعیفتری به نام پاد استوکس با فرکانس νa ظاهر شوند.
نوارهای رامان
اختلاف انرژی νs - ν0 یا νaν0 - به صورت انرژی ارتعاشی یا دورانی مولکول داده (یا از آن گرفته) میشود. از این روش میتوان برای بررسی مدهای ارتعاشی مولکولی که منجر به جذب فروسرخ نمیشوند، استفاده کرد. شرط وجود ظهور طیف رامان این است که مولکول دارای قابلیت قطبی شدنی باشد که به حالت ارتعاشی یا دورانی بستگی دارد. در مولکولهای دو اتمی این وضع همیشه هست و طیف رامان شامل یک نوار استوکس و یک نوار پاداستوکس در فرکانسهای ω ± است که ساختار دورانی هر کدام بوسیله قاعده گزینش به صورت 2± و 0ΔJ= تعیین میشود.
|
طیف رامان دورانی
ساختمان دورانی در حول خط تابشی ظاهر میشود، مولکولهای چند اتمی طیفهای رامان دورانی خالص نیز دارند (جز برای مولکولهای نوع تقارن کروی) معمولا بعضی از مدهای ارتعاشی رامان فعال بوده و بعضی چنین نیستند. در موارد مشخصی قواعد مربوط به ظهور طیفهای فروسرخ و رامان بطور متقابل منحصر به فرد هستند. در حقیقت ، نوع تقارن در مولکول را میتوان صرفا از وجود یکی از طیف های رامان یا فروسرخ یا هر دوی آنها نتیجه گرفت.
لطفا به ادامه مراجعه فرمایید:
اوربیتالهای مولکولی
در مولکولها، مانند اتمهای مجزا، الکترونها اوربیتال ها را بر طبق همان قواعد پیشین پر میکنند. این اوربیتال های مولکولی را به صورت متمرکز در اطراف چندین هسته و شاید در بر گیرنده ی تمامی مولکول، در نظر میگیرند؛ توزیع هسته ها و الکترونها به صورتی است که پایدارترین مولکول ممکن از آن نتیجه شود.
برای آنکه معادله های پیچیده ی ریاضی بیشتر قابل استفاده باشند، معمولاً از دو فرض ساده کننده بهره میگیرند:(الف)هر جفت الکترون اساساً در نزدیکی دو هسته مستقرمی شود، و(ب) شکل این اوربیتال های مولکولی مستقر و موضع آنها نسبت به یکدیگر، به صورت ساده، وابسته به شکل و موضع اوربیتال های اتمی در اتمهای سازنده است.
تصور اوربیتالهای مستقر- یا آنچه که ما آن را اوربیتالهای پیوندی می نامیم- ظاهراً فکر بدی نیست زیرا این روش تقریب، از دیدگاه ریاضی، در مورد بیشتر مولکولها (ولی نه همه ی آنها) موفقیت آمیز است.
افزون بر این این تصور همردیف با مفهوم کلاسیک شیمیدانها در مورد پیوند به عنوان نیرویی که بین دو اتم وجود دارد و تقریباً مستقل از باقی مانده مولکول عمل میکند، قرار میگیرد؛ تصادفی نیست که این مفهوم به گونه ای شگفت انگیز به مدت یک سال کارایی مطلوب داشته است. نهایتاً مولکولهایی که بطور استثنایی، فرمولهای کلاسیک در مورد آنها کارایی ندارد، همان مولکولهایی هستند که روش اوربیتال مولکولی مستقر نیز در مورد آنها کارایی ندارد.
فرض دوم بسیار منطقی است. این فرض به اندازه ای سودمند بوده که هر زمان لازم باشد، اوربیتالهای اتمی از انواع معین اختراع می شود تا این فرض همچنان معتبر باقی بماند.
 منبع:
منبع: